
Pathomechanisms of Diabetic Kidney Disease

1
Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, CA 90095, USA
2
College of Medicine, Charles R Drew University of Medicine and Science, Los Angeles, CA 90059, USA
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2023, 12(23), 7349; https://doi.org/10.3390/jcm12237349
Submission received: 10 October 2023
/
Revised: 15 November 2023
/
Accepted: 22 November 2023
/
Published: 27 November 2023
(This article belongs to the Special Issue Editorial Board Members’ Collection Series: Chronic Kidney Disease in Diabetes)
Abstract
:The worldwide occurrence of diabetic kidney disease (DKD) is swiftly rising, primarily attributed to the growing population of individuals affected by type 2 diabetes. This surge has been transformed into a substantial global concern, placing additional strain on healthcare systems already grappling with significant demands. The pathogenesis of DKD is intricate, originating with hyperglycemia, which triggers various mechanisms and pathways: metabolic, hemodynamic, inflammatory, and fibrotic which ultimately lead to renal damage. Within each pathway, several mediators contribute to the development of renal structural and functional changes. Some of these mediators, such as inflammatory cytokines, reactive oxygen species, and transforming growth factor β are shared among the different pathways, leading to significant overlap and interaction between them. While current treatment options for DKD have shown advancement over previous strategies, their effectiveness remains somewhat constrained as patients still experience residual risk of disease progression. Therefore, a comprehensive grasp of the molecular mechanisms underlying the onset and progression of DKD is imperative for the continued creation of novel and groundbreaking therapies for this condition. In this review, we discuss the current achievements in fundamental research, with a particular emphasis on individual factors and recent developments in DKD treatment.
1. Introduction
With the rising global prevalence of diabetes mellitus (DM) [1,2], diabetic kidney disease (DKD) remains the leading cause of chronic kidney disease (CKD), often progressing to kidney failure requiring replacement therapy in the form of dialysis or kidney transplantation [3,4,5]. Additionally, DKD is closely associated with heightened cardiovascular risks, including coronary artery disease, heart failure, sudden cardiac death, and increased morbidity and mortality [6,7]. The impact of DKD on both individuals with DM and healthcare systems is substantial. The development and progression of DKD are believed to stem from the complex interplay of metabolic, hemodynamic, inflammatory, and fibrotic factors, which are frequently disrupted in DM [8,9,10]. Dysfunction in these factors may be interlinked, influencing gene regulation, activating transcription factors, and affecting molecular pathways [8,10]. These interactions lead to functional and structural changes culminating in the clinical manifestations of DKD, characterized by escalating albuminuria and declining renal function [8]. While conventional approaches like optimal blood pressure control through renin–angiotensin–aldosterone system (RAAS) blockade and glycemic control have demonstrated efficacy in slowing down the progression of DKD, they do not halt or reverse the condition [11]. Therapies directed at pathways associated with metabolic changes, renal inflammation, fibrosis, and oxidative stress have displayed notable benefits in animal models. Consequently, several of these agents have undergone examination in clinical trials involving human subjects, yielding varied outcomes [11]. This review concentrates on several of the pathways and factors contributing to renal injury associated with DKD, drawing from a range of experimental nephropathy models, including published studies conducted by the author’s research group. For instance, our laboratory initially demonstrated for the first time that osteopontin (OPN) modulates angiotensin II (AngII)-induced inflammation, oxidative stress, and fibrosis in OPN-null mice in the heart [12] and then in the kidney [13]. We later showed that OPN plays a critical role in the development of DKD [14]. Our research was then used as the basis to reveal that the proteolytically cleaved fragment of OPN, known as N-terminal OPN (nt-OPN), exhibits a more substantial role in this pathology [15], and these findings are discussed. In addition to a description of established and novel pathways to DKD, we discuss existing therapies that target specific pathways involved in DKD.
Factors That Promote DM and DKD
The development of DKD involves multiple factors, encompassing structural, metabolic, hemodynamic, inflammatory, and fibrotic processes in addition to genetic factors. These contribute collectively to the gradual deterioration of kidney function (Figure 1).
2. Metabolic Factors
The early alterations in DKD are initiated by metabolic factors, particularly elevated blood glucose levels or hyperglycemia [16]. The harm caused by hyperglycemia can arise from direct tissue modifications or byproducts generated during glucose metabolism [16]. A visual representation of the disrupted metabolic pathways driving DKD development in individuals with DM is depicted in Figure 2 and discussed below.
Elevated blood glucose levels trigger the activation of pathways such as hexosamine, polyol, advanced glycation end products (AGE), and protein kinase C (PKC) [17]. This activation results in increased production of reactive oxygen species (ROS) and higher levels of mitogen-activated protein kinase (MAPK), JAK signal transducers and activators of transcription, and NF-κB [18]. These factors collectively contribute to the development of inflammation and fibrosis. MAPK is associated with the production of the extracellular matrix (ECM) and injury to podocytes [19]. NF-κB prompts the generation of adhesion molecules and cytokines, including monocyte chemoattractant protein (MCP-1), IL-6, and tissue necrosis factor (TNF)-α [20]. Additionally, ROS directly inflicts damage on cellular structures by oxidizing various lipids, nucleic acids, and proteins [21]. Here we discuss each of these pathways in detail.
2.1. Pathways Affected by Metabolic Disturbances in Hyperglycemia
2.1.1. PKC Pathway
PKC is an intracellular signaling molecule that has the potential to slow down, or even halt the advancement of diabetic complications, including DKD [22,23]. The PKC family encompasses several isoforms including: α, β I, β II, γ, and δ [24], and the role of these isoforms in the development of DKD has been further clarified through the use of experimental rodent models [24]. Also, the elevated expression of mRNAs associated with the PKC-MAPK pathway is linked to glomerular lesions in patients with diabetic nephropathy (DN) [25]. Findings indicate that the PKC pathway is associated with the advancement of DKD by narrowing small blood vessels in the kidney and thus affecting the function of glomeruli [26]. In the mouse model of DM, phenylephrine-induced contraction of interlobar arteries is significantly augmented, inducing interlobar artery dysfunction which further diminishes blood flow within the glomerulus, facilitating the progression of DKD. Treatment with rottlerin, a calcium-independent PKC inhibitor, mitigates excessive basal contraction [27,28]. Similar findings were observed in diabetic Zucker rats, where the nonselective PKC agonist phorbol-12,13 dibutyrate reduced renal cortical blood flow and raised mean arterial pressure [29]. These outcomes highlight the association of PKC activation with decreased blood flow, heightened renal perfusion pressure, glomerulosclerosis, and reduced glomerular filtration.
The role of PKC in vascular permeability in DKD is particularly pronounced in the glomeruli, and PKC-α and PKC-β have emerged as significant players in maintaining glomerular filtration function [30,31]. Studies revealed that PKC-α and PKC-β were upregulated in diabetic mice, and the deletion of NOX4 reversed their overexpression and subsequently normalized nephrin expression [31], which is crucial for podocytes in maintaining slit diaphragm integrity for optimal filtration. Further, mediation of the ras homolog gene family, member A (RhoA) downstream of C3aR in endothelial cells by PKC resulted in kidney damage and increased blood vessel permeability [32].
In DKD, expansion of the glomerular basement membrane (GBM) and the accumulation of ECM are notable features [33]. In this pathology, the elevated activation of PKC was observed along with the increased production of ECM proteins such as fibronectin, laminin, and types I, III, and IV collagen in renal glomerular mesangial cells [34,35,36]. This effect is replicated by PKC activators like phorbol 12-myristate 13-acetate and oleoyl acetyl glycerol, a cell-permeable diacylglycerol analog [34]. The role of PKC activation in ECM protein synthesis and ECM degradation is further underscored in various studies. For instance, hyperglycemia promotes the secretion of hyaluronan (HA), a key ECM element, and PKC-β inhibition curbs HA secretion by reducing HAS2 mRNA expression [37]. A specific PKC inhibitor (GF 109203X) counteracts hyperglycemia-induced elevation in collagenous and total ECM protein synthesis and reduces human endothelial cell gelatinase activity [38]. PKC-α and PKC-β activation is also elevated in mesangial cells under high glucose concentrations, paralleling fibronectin, and IV collagen synthesis [39,40]. LY333531, a specific PKC-β inhibitor, halts ECM component expression in mesangial cells in hyperglycemia [40]. Global PKC-α/PKC-β double knockout mice exhibit diminished ECM component expression alongside reduced albuminuria development [24]. PKC-δ-/- and PKC-ε-/- streptozotocin (STZ)-induced diabetic mice, and mice with inhibited PKC-δ, reveal reduced ECM protein synthesis in mesangial cells [41,42,43]. Notably, PKC-ε-/- mice exhibit a profibrotic phenotype solely in the kidney [42]. The activation of PKC-δ, instead of PKC-α or PKC-β, is heightened in mesangial cells under high glucose conditions [43]. The membrane association of PKC-ζ, demonstrated to be hindered by rosiglitazone, is linked to reduced collagen IV expression in mesangial cells [44].
Transforming growth factor β1 (TGF-β1) plays a significant role in the accumulation of GBM and ECM in DKD [45,46] along with heightened activation of PKC [22,47,48]. Inhibition of PKC-α and PKC-β leads to a reduction in the expression of TGF-β1 and connective tissue growth factor (CTGF) in both glomerular endothelial cells and mesangial cells [22,39,40,47,49,50]. Mice with a knockout of Akr1b3 show inhibited PKC activation, resulting in decreased ECM accumulation and glomerular hypertrophy [51]. Homologous genes NOX2, NOX4, and NOX5 suppress the activation of PKC-α and PKC-β, leading to reduced levels of ROS, TGF-β1 expression, and MCP-1 in conditions of hyperglycemia. This, in turn, helps prevent ECM accumulation and thickening of the GBM in mesangial cells and podocytes [52,53,54].
Extracellular signal-regulated kinase (ERK) also plays a role in ECM accumulation. PKC-δ expression is increased under high glucose conditions, and its inhibition through rottlerin suppresses ERK expression and TGF-β1 responsiveness, blocking the fibrotic response in mesangial cells [55]. Conversely, PKC-β inhibition reduces ERK1 and ERK2 expression in mesangial cells [56]. Moreover, the involvement of phospholipase C-γ1 (PLC-γ1) in DKD development is highlighted; PLC-γ1 inhibition curbs PKC-β-induced protein kinase B (AKT) S473 phosphorylation and prevents collagen I upregulation [57]. In summary, PKC overexpression is implicated in ECM accumulation, primarily arising from mesangial cells; however, findings on PKC isoforms yield mixed results [24,43,58].
These findings indicate that the pursuit of a therapeutic approach involving isoform-specific inhibitors for targeting the PKC pathway holds promise. However, robust support for this potential treatment avenue necessitates well-designed, large-scale, and long-term clinical studies.
2.1.2. AGEs/Receptor for Advanced Glycation End Products (RAGE) in DM
AGEs are enduring post-translational protein modifications that arise from the spontaneous interaction with glucose and associated metabolites. The AGEs cause harmful effects in two ways: firstly, by directly trapping and linking proteins together, and secondly, by attaching to a receptor on the cell surface [59,60]. Although AGEs can interact with various receptors, the precise interactions and their roles in cellular responses are not fully understood [61,62]. AGEs can influence cell functions by binding to toll-like receptors, scavenger receptors, G-protein-coupled receptors, and pattern recognition receptors [61,63]. Among these, the most crucial cell surface receptor for AGEs is RAGE, a member of the immunoglobulin superfamily. It was initially identified due to its ability to bind with AGEs [64,65]. Notably, RAGE is unique in its capacity to recognize three-dimensional structures rather than specific amino acid sequences. RAGE is considered a pattern-recognition receptor because of its ability to identify the structural characteristics of its ligands [64,65,66].
The full-length RAGE (fl-RAGE) protein, the most prevalent form, is composed of three domains: an extracellular domain with a V-type N-terminal domain and two C-type (C1 and C2) immunoglobulin domains, a transmembrane domain, and a cytosolic domain rich in charged amino acids [64,65]. The extracellular V-type domain primarily interacts with potential ligands outside the cell, while the cytoplasmic tail is crucial for intracellular signaling and serves as a scaffold for initiating signal transduction [67]. Also, the primary RAGE transcript undergoes alternative splicing and proteolytic cleavage of fl-RAGE to produce truncated RAGE isoforms, a process governed by unknown pathways [68,69,70]. The activation of RAGE by AGEs leads to an increase in RAGE receptor expression, creating a positive feedback loop where ligand-stimulated RAGE amplifies and sustains its own activity [71,72].
In DM, elevated circulating AGEs interact with its receptor, RAGE, and induce downstream signaling molecules, including MAPK, p38, stress-activated PKC-Jun N-terminal kinase (SAPK/JNK), Ras-mediated ERK1/2, and the JAK/STAT pathway. These pathways subsequently lead to the prolonged activation of transcription factors such as NF-κB, STAT3, HIF-1α, and AP-1 [62,73,74]. Hence, intervening in DKD by targeting RAGE along with its ligands to mitigate oxidative stress and chronic inflammation is viewed as an additional strategy. Nevertheless, a clinical trial involving an advanced AGE inhibitor, while demonstrating protective effects against kidney injury [75], was halted due to severe side effects [76].
2.1.3. Hexosamine Pathway
The hexosamine biosynthetic pathway is thought to be involved in the progression of insulin resistance and the emergence of vascular complications in DM. It holds significance in the synthesis of proteoglycans, glycolipids, and glycoproteins [77]. This pathway involves converting fructose-6-phosphate (fruc-6-P) into glucosamine-6-phosphate (glucN-6-P) using glutamine as the amino donor, mediated by the rate-limiting enzyme, glutamine: fructose-6-phosphate-amidotransferase (GFAT). Subsequently, glucN-6-P is rapidly utilized in generating uridine-5-diphosphate-N-acetylglucosamine (UDP-N-acetylglucosamine), a precursor required for synthesizing amino sugars needed for glycoproteins, proteoglycans, glycosaminoglycans, and glycolipids [77,78,79]. Elevated blood sugar levels foster DM complications by increasing fruc-6-P concentration, driving it into the hexosamine biosynthetic pathway [80,81]. Concomitantly, heightened glucose concentrations induce metabolic pathways that culminate in the release of cytokines such as TGF-β, ICAM-1, VCAM-1, TNF-α, CTGF, and plasminogen activator inhibitor (PAI)-1, which play roles in diverse diabetic complications [82,83]. For instance, TGF-β1 significantly contributes to DKD [77]. The critical role of PAI-1 in DKD was demonstrated in our own work in which PAI-1 was shown to regulate TGF-β1 expression, and deletion of PAI-1 reduced TGF-β1 and retarded the development of DN [84]. Cellular glucose uptake also channels a relatively larger portion of glucose to glycogenesis, glycolysis, and pentose phosphate metabolism. Additionally, about 2–3% of glucose molecules are directed into the hexosamine biosynthetic pathway [83,85]. Blocking the hyperglycemia-induced transcription of cytokines is achieved by inhibiting the rate-limiting enzyme, GFAT, which effectively prevents potential diabetic complications arising from this pathway [77,80,86]. It was demonstrated that the heightened production of mitochondrial superoxide due to hyperglycemia leads to an elevation in hexosamine synthesis and the O-glycosylation of specificity protein 1 (Sp1). This, in turn, triggers the activation of genes that play a role in the development of diabetic complications [87].
2.1.4. Polyol Pathway
The polyol pathway involves two enzymatic reactions [88,89]. The initial reaction is the reduction of glucose to sorbitol, facilitated by the enzyme aldose reductase (AR). This step is considered the rate-limiting process in the pathway and results in the conversion of NADPH to NADP+ [89,90]. The subsequent reaction transforms sorbitol into fructose, catalyzed by sorbitol dehydrogenase, generating NADH from NAD+. Thus, the end products of the polyol pathway include sorbitol, fructose, and NADH [89]. Therefore, in DM, this pathway is believed to be a primary contributor to the redox imbalance between NADH and NAD+ [89,91,92].
In a murine model of STZ-induced DM, the absence of the AR gene significantly improved the progression of early DKD indicators [51]. In this study with diabetic AR-null mice, there was complete inhibition of DM-induced ECM build-up and excessive collagen IV production. Additionally, deficiency of AR led to a complete or partial blockade of the DM-induced activation of renal cortical PKC, TGF-β1, and glomerular hypertrophy. In diabetic AR-null mice, there was a significant decrease in urine albumin excretion [51]. Studies have revealed the potential of AR inhibitors (like pyrogallol) in alleviating DM and a variety of AR inhibitors have undergone testing and assessment [93]. In contrast to the experimental evidence, AR inhibitors only have a partial effect in preventing DKD in patients [92].
In hyperglycemic conditions, the activation of the polyol pathway results in an increase in fructose levels, potentially worsening DM, and its related complications [89,94]. Fructose has dual effects: it can chemically glycate proteins, leading to their dysfunction [95]; additionally, its metabolism by fructokinase, which consumes ATP, bypasses the regulatory mechanisms of the glycolytic pathway [96,97]. This can lead to an overproduction of acetyl-CoA and depletion of ATP, further contributing to protein functional impairment [98,99].
Moreover, the kidney, being abundant in mitochondria [100], implicates the redox reactions and energy metabolism within these organelles in the onset of metabolic disorders in the kidney, including DKD [101]. On a molecular level, altered glucose metabolism leads to a notable redox imbalance in the NADH/NAD+ ratio [102,103], which may emerge as a distinctive mechanism contributing to diabetic kidney injury [104,105]. This is due to the fact that electrons generated from the breakdown of glucose and other nutrients like fatty acids and amino acids are retained in NADH, utilizing NAD+ as the electron acceptor [106,107]. Consequently, a prominent characteristic of DM is an excess of NADH and a deficiency of NAD+ [89,103]. Several pathways have been investigated for their involvement in NAD consumption. These include the Poly ADP Ribosylation Pathway [108], Sirtuins Pathway [109], CD38 Pathway, and NAD Kinase Pathway [110]. The significance of the Poly ADP Ribosylation Pathway in the development of DM has been established through studies in murine models, where the absence of this enzyme prevents the onset of DM [111]. This highlights the harmful consequences of NAD+ depletion in diabetes. Additionally, research has demonstrated that CD38-driven NAD+ deficiency is responsible for organ fibrosis and dysfunction in the kidneys of diabetic individuals [112].
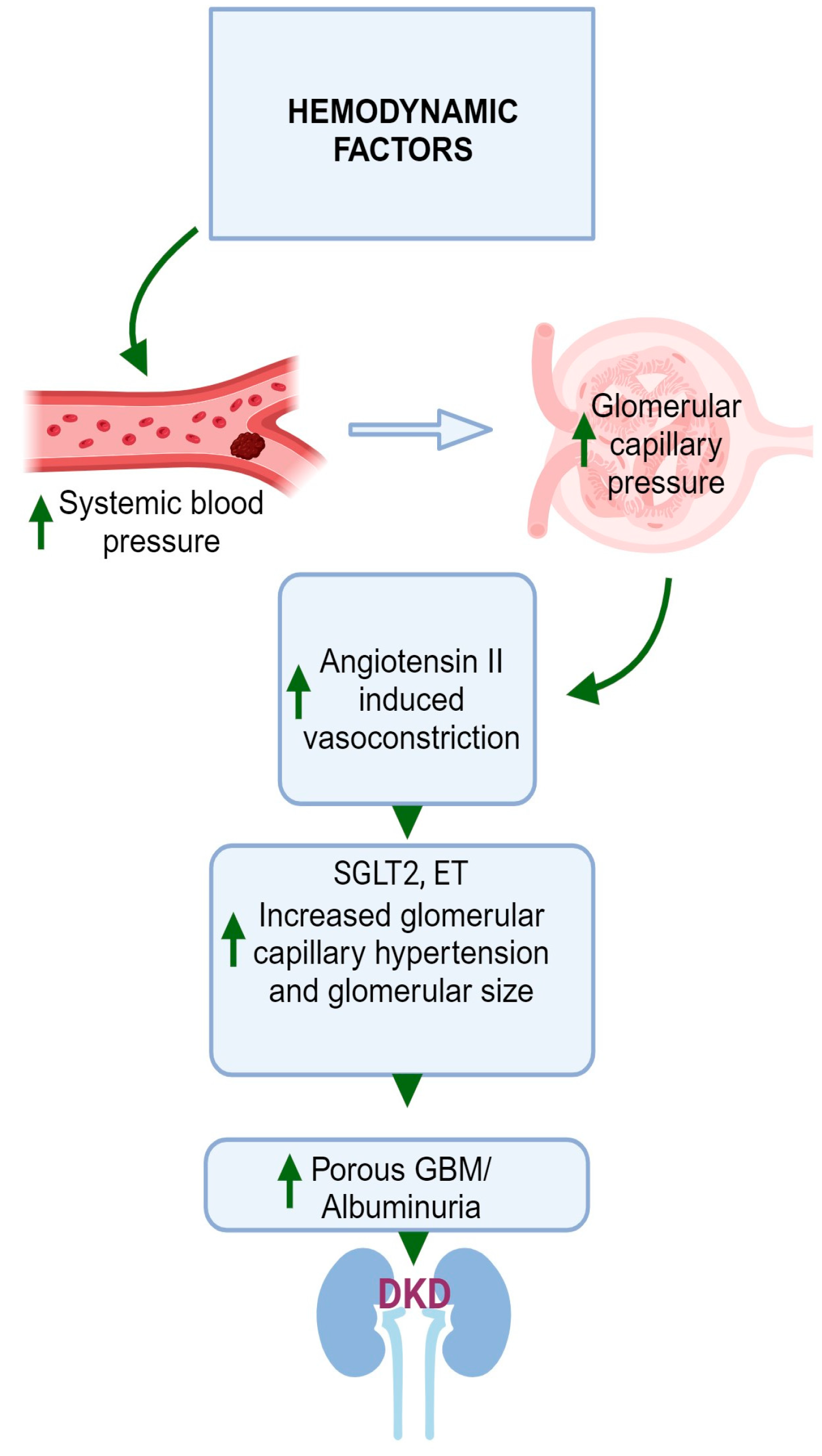
3. Hemodynamic Factors
DM may also lead to hemodynamic effects, which include increased systemic blood pressure [113] and increased intraglomerular pressure [114]. The elevation in glomerular capillary pressure gives rise to an increase in the glomerular filtration rate (GFR) of a single nephron, a phenomenon referred to as hyperfiltration [115,116,117]. In DKD, glomerular hyperfiltration is the first stage in the pathogenesis, leading to progressive albuminuria, declining GFR, and finally kidney failure or end-stage renal disease (ESRD), although the mechanism is not fully understood [115]. The escalation in intraglomerular pressure arises from heightened tone in the efferent arteriole coupled with a reduction in tone in the afferent arteriole [117]. The exact mechanism behind this process is not definitively established, yet two main theories have emerged.
One perspective posits that hyperfiltration is driven by molecules present in the circulation that exert their effects predominantly within the glomerulus [118]. Multiple mediators have been suggested as agents that could amplify intraglomerular pressure by increasing tone in the efferent arteriole and diminishing tone in the afferent arteriole. Elevated resistance in the efferent arteriole can arise due to heightened levels of AngII, thromboxane A2, endothelin (ET) 1, and ROS [117]. Conversely, diminished resistance in the afferent arteriole can be initiated by a decrease in the availability of nitric oxide, an increase in cyclooxygenase-2 prostanoids, activation of the kallikrein-kinin system, atrial natriuretic peptide, angiotensin 1–7, and an increase in insulin [117].
However, another perspective suggests that tubular mechanisms play a more pivotal role in driving intraglomerular hypertension [119]. The activation of glucose transport pathways in the proximal tubule at the early stages of DM triggers enhanced reabsorption of both glucose and sodium in the proximal nephron [119]. Consequently, the delivery of sodium to the distal nephron decreases. This prompts a response known as tubuloglomerular feedback, leading to dilation of the afferent arteriole and constriction of the efferent arteriole [119]. The sodium–glucose cotransporter 2 (SGLT2) is a postulated mechanism of glomerular hyperfiltration. SGLT2 increases the reabsorption of glucose in the proximal tubules, thereby reducing the delivery of sodium chloride to the macula densa [120]. As a result, tubuloglomerular feedback is reduced, afferent arterioles are dilated, and AngII is increased in efferent arterioles, resulting in vasoconstriction [121,122]. These effects increase glomerular perfusion and intraglomerular pressure, leading to glomerular hyperfiltration. Moreover, an increase in insulin on its own can enhance sodium and glucose transport in the proximal tubule, thereby inciting tubuloglomerular feedback. As highlighted earlier, insulin can also directly reduce tone in the afferent arteriole. As a result, insulin can exert both direct and indirect influences that contribute to the occurrence of hyperfiltration. Figure 3 illustrates a graphical depiction of the altered hemodynamic factors contributing to the development of DKD in individuals with DM.
3.1. RAAS Pathway
The intrarenal hemodynamic function is significantly impacted by the RAAS [123]. This system heightens oxidative stress and triggers pro-inflammatory pathways. These processes lead to glomerular hypertrophy, a feature that manifests in the early stages of DKD and marks the initiation of a profibrotic cascade [124]. The favorable effects of RAAS blockers on retarding the progression of DKD have been extensively documented, as evidenced by numerous studies [125]. This effect holds true despite the presence of a relatively low systemic renin state in individuals with DKD. This phenomenon is believed to be a manifestation of the activated local renin system within the kidneys or an increased sensitivity to AngII at the intrarenal level [126]. The varying stages of kidney disease and diverse assessment methods used (including measuring renin activity, protein levels, RNA expression, serum potassium, and bicarbonate concentrations, and employing techniques like immunohistochemistry and fluorescence) have resulted in conflicting clinical data regarding the measurement of RAAS activation in DKD [127,128]. Conversely, findings from experimental models have consistently demonstrated elevated RAAS activation in DKD [129,130,131,132]. This activation of the RAAS encompasses all elements and stages of the cascade, and it occurs in a localized manner, operating in a paracrine fashion. The RAAS cascade commences with the synthesis of prorenin within the juxtaglomerular cells, which is subsequently cleaved into renin [133]. Renin then catalyzes the cleavage of angiotensinogen (AGT) to generate angiotensin I (AngI). This AngI is further transformed by the angiotensin-converting enzyme (ACE) into the octapeptide known as AngII.
RAAS blockade has been observed to have favorable effects on renal outcomes predominantly in placebo-controlled clinical trials [125,134]; however, the favorable effect may potentially be explained by the blood pressure-lowering effect of RAAS blockade [135]. Significant trials have demonstrated distinct advantages of AngII receptor blockers (ARB) for individuals with DKD [136,137]. It is theoretically considered that dual blockade is more effective than single blockade because ACEIs and ARBs act on different sites within the RAAS [138]. Early studies suggested a combination of ACE inhibitors and ARB may provide additional benefits in diabetic nephropathy [139,140], however, this combination is not clinically recommended due to complications of hyperkalemia and acute kidney injury.
3.2. Prorenin and Renin
In individuals with DM, approximately 95% of circulating renin is in the form of prorenin. The precise mechanism through which prorenin contributes to the development of DKD is not well known [141]. Prorenin binds to the prorenin receptor (PRR) and, independently of AngII, initiates intracellular signaling. This signaling leads to the activation of mitogen-activated protein (MAP) kinases ERK1/2, resulting in the upregulation of proteins such as TGF-β1, PAI-1, collagens, fibronectin, and cyclooxygenase-2 [142,143,144,145,146]. This suggests that elevated prorenin levels might contribute to the progression of DKD by stimulating PRR and prompting the synthesis of pro-fibrotic proteins [133]. Notably, in experiments with transgenic rats that overexpress human PRR, these rats develop proteinuria and exhibit a gradual onset of glomerulosclerosis and kidney damage independent of AngII [129]. Intriguingly, inducing overexpression of prorenin alone does not lead to glomerulosclerosis, hinting that prorenin may enhance fibrosis rather than directly causing it [130,147]. Additionally, prorenin plays a role in generating AngI. When prorenin binds to PRR, it induces a conformational change that involves the unfolding of the peptide from the enzymatic cleft, making the cleft accessible to AGT and allowing the generation of AngI [148,149]. In patients with DM, elevated plasma prorenin levels were observed [150], alongside reduced plasma renin levels compared to normal healthy subjects [126]. The coexistence of elevated prorenin levels and heightened renal PRR expression in DM [151] implied a potential involvement of this receptor in the development of DKD.
In the early stages of DM, there is a notable increase in the expression of renin mRNA within the proximal tubule [131]. Beyond its traditional role in enhancing the synthesis of AngII, renin has a direct effect on stimulating the production of TGFβ, a cytokine associated with fibrosis [143]. Renin binds to its specific receptor on the cell surface of mesangial cells, resulting in cell hypertrophy and an increased efficiency of AGT cleavage by renin. This process unmasks the catalytic activity of prorenin [152]. Interestingly, the renin receptor has also been found in the sub-endothelium of renal arteries, suggesting that renin has a novel receptor-mediated action that could potentially contribute to renal fibrosis [153]. Moreover, in podocytes, elevated glucose levels have been demonstrated to lead to increased AngII generation. This occurs by boosting renin mRNA expression, accompanied by a simultaneous increase in the PRR levels, ultimately enhancing the conversion of AGT to Ang I [154].
3.3. ACE
The most compelling evidence supporting the existence of intrarenal renin production and activity in DM comes from animal models of spontaneous DM or those induced by STZ [131]. Interestingly, despite having either suppressed or normal levels of plasma renin activity, DM leads to increased renin mRNA and protein levels within the kidneys, particularly in the proximal tubules [131]. In rats with DM, there is a notable reduction in total renal ACE activity, accompanied by a specific redistribution of ACE within the diabetic kidneys [155]. While ACE activity in the proximal tubules is decreased, staining intensity for ACE is intensified in diabetic glomeruli and the renal vasculature. This observation suggests a potential role for glomerular ACE in mediating nephron injury, potentially by enhancing the local formation of AngII within the glomerulus [155].
3.4. AngII
As mentioned earlier, DKD is associated with an upsurge in intrarenal AngII generation, despite the systemic suppression of the RAAS. The adverse effects of this elevation in AngII extend beyond hemodynamic alterations and encompass insulin resistance, cell growth stimulation, and damage to the tubules. One of the pivotal roles of AngII in DKD is its association with volume expansion achieved through water and sodium reabsorption.
AngII activates the luminal membrane Na+–H+ antiporter by stimulating an inhibitory G protein, which in turn reduces cyclic AMP generation. This reduction minimizes the typical suppressive influence of cyclic AMP on Na+–H+ exchange [68,156]. AngII also stimulates phosphatidylinositol turnover, leading to the production of PKC [156,157]. Moreover, it enhances the secretion of aldosterone from the adrenal cortex, further promoting sodium transport in the cortical collecting tubule [158]. In the proximal tubule, AngII inhibits proteinase activity and induces mesangial cell expansion by decreasing the activity of the plasminogen activator. Additionally, AngII leads to the upregulation of TGF-β1 and the release of vascular endothelial growth factor from glomerular epithelial and mesangial cells, both of which contribute to mesangial matrix expansion [159]. Renal fibroblasts express AngII type 1 (ATI) receptors and respond to AngII stimulation by proliferating, expanding the matrix, and synthesizing fibronectin, primarily through a TGF-β-dependent mechanism [160].
Microinflammation within the glomeruli and tubulointerstitial regions, followed by the expansion of the ECM, are shared pathways in the progression of DKD. AngII activates inflammatory cells directly, causing chemotaxis, including the release of OPN, RANTES, and other proinflammatory mediators like MCP-1 and TGF-β. It also triggers the activation of various intracellular signaling pathways, including PKC, protein tyrosine kinases, MAPK, ERK, c-Jun amino-terminal kinase (JNK), p38 MAP kinase (p38 MAPK), and the activator protein-1 (AP-1). These factors are implicated in processes such as proliferation, differentiation, fibrosis, and inflammation [160].
Furthermore, AngII has been linked to insulin resistance [161]. It hinders insulin-mediated GLUT4 translocation in skeletal muscle by transiently activating ERK1/2, which inhibits insulin receptor substrate ½ (IRS-1/2). Additionally, AngII directly inhibits insulin sensitivity through nitration of AKT and induces tyrosine phosphorylation of IRS-1 via Janus kinase 2, associated with AT1 receptor stimulation. This phosphorylation attenuates insulin-induced activation of phosphatidylinositol-3-kinase, ultimately leading to reduced insulin sensitivity [162]. Interestingly, treatment with the AngII receptor blocker candesartan increases renal insulin receptor expression in STZ-induced diabetic rat models and insulin-resistant Zucker rats, regardless of insulin levels [163].
In human DN, components of the RAS undergo alterations in the kidney, revealing increased local AngII production, activation of tubular cells, and the induction of proinflammatory markers. This indicates that AngII plays a role in the renal inflammatory process, potentially providing insight into the molecular mechanisms responsible for the beneficial effects of RAS blockade [164].
4. Inflammatory Factors
In the progression of DKD, several interconnected pathways contribute to disease development, and inflammation serves as a crossroad in its pathogenesis. DM initiates inflammatory processes mediated by several factors like oxidative stress, AGEs, obesity, ischemia, and cellular damage [165]. These processes collectively generate inflammatory molecules, including NF-κB, caspases associated with the NLR family pyrin domain containing 3 (NLRP3) inflammasome, TNF-α, IL-1β, IL-6, and IL-18.
Araújo et al. examined the expression levels of various cytokines and chemokines, including CCL11, macrophage inflammatory protein-1α, IL-8, IL-4, IL-10, TNF-α, TNFR1, IL-1β, and IL-6, in renal biopsies taken from patients diagnosed with DN. Their results demonstrated an increased expression of cytokines and chemokines in DN, specifically highlighting elevated levels of IL-6, IL-1β, IL-4, and CCL11. The study concluded that CCL11 might exert a significant influence on the progression of interstitial inflammation in DN and contribute to the decline in estimated GFR in these patients [166].
According to a separate study utilizing renal biopsies, it has been suggested that in DN, the primary inflammatory agent driving renal inflammation is IL-1α, rather than IL-1β, which is released by renal tubular cells [167]. Additionally, levels of IL-1α in both the urine and plasma of DM patients have been linked to markers indicating injury in podocytes and proximal tubular epithelial cells [168].
Similarly, urinary IL-6 could be a valuable indicator for identifying DKD in DM patients, even in cases where there is no detectable urinary albumin excretion [169]. Elevated levels of IL-6 in the serum have been observed in individuals with DKD [170] and were linked with GBM thickness [171]. Also, the expression of IL-6 mRNA in glomerular and interstitial cells in individuals with DKD was associated with mesangial proliferation causing renal injury [172]. In patients with DM, urinary IL-6 was also connected to the progression of DKD [173].
Furthermore, TNF-α serves as a crucial transcriptional regulator for components of the NLRP3 inflammasome [174,175]. In patients with DKD, elevated levels of serum TNF-α and its receptor have been observed, and they are indicative of both renal decline and the likelihood of ESRD occurrence [176,177]. It has been demonstrated that inhibiting TNF-α can notably alleviate glomerular lesions in murine models of DM [178]. The TNF-α inhibition provides protective effects against tubulointerstitial nephritis by suppressing the NLRP3 inflammasome in DN rats [179].
The increase in AGEs has been directly associated with an elevated expression of NLRP3-related proteins, which have been proposed as mediators of CKD, potentially activating mesangial cells [180]. NLRP3-related proteins are present in macrophages and inflammasomes and have been associated with various inflammatory disorders. In several mouse models, reducing NLRP3 activity has been shown to slow down the progression of CKD in a dose-dependent manner. Additionally, inflammation can lead to the infiltration of neutrophils and macrophages, oxidation of lipoproteins, and deposition of immune complexes [181]. This persistent inflammation also results in increased production and deposition of amyloid A protein, which can serve as a marker of disease progression [182].
Moreover, the activation of the complement system significantly influences the progression of DKD. The activation of the complement system in DKD has been linked to mannose-binding lectins and the ficolin-associated activation of the lectin pathway within the complement cascade. Hyperglycemia results in higher levels of glycan and galactosamine-bound substances that are recognized by these receptors, leading to complement activation [183]. Ongoing research may yield new complement inhibitors that offer benefits to DKD patients; however, careful monitoring is crucial to evaluate any impact on susceptibility to infections or immune complex diseases.
In summary, DKD involves multiple intertwined pathways, with inflammation also playing a pivotal role in its development (Figure 4). This inflammation is initiated by various factors associated with DM and results in the production of inflammatory molecules and activation of the complement system, both of which contribute to the progression of the disease.
5. Fibrotic Factors
Renal fibrosis refers to an excessive buildup of ECM in the kidney, which can manifest as either glomerulosclerosis or tubulointerstitial fibrosis [184,185]. The main components of this fibrotic ECM consist of collagens I and III, known as fibrillar collagens, along with glycoproteins and proteoglycans [186]. The extent of fibrosis is closely linked to the decline in renal function and the progression towards ESRD [187]. Myofibroblasts play a pivotal role in facilitating ECM deposition due to their robust capacity to produce ECMs like collagens, thus contributing to the advancement of renal fibrosis [188]. In individuals with DM, there is a notable infiltration of activated myofibroblasts, a phenomenon generally not observed in a healthy physiological state [189]. However, there is ongoing debate regarding the role of myofibroblasts, particularly the process of epithelial to mesenchymal transition (EMT) where tubular epithelial cells transform into myofibroblasts in DKD [190]. Renal fibrosis constitutes a crucial pathological alteration in DKD and significantly elevates the mortality rate among late-stage DKD patients. The major signaling pathways implicated in renal fibrosis encompass the TGF-β, MAPK, Wnt/β-catenin, PI3K/AKT, JAK/STAT, and Notch pathways (Figure 5). Each of these pathways exerts a substantial influence on the buildup of ECM, the expression of collagen and fibronectin, as well as the secretion of other pertinent proteins [191].
5.1. TGF-β Signaling Pathway
It is widely recognized that TGF-β stands as the most significant profibrotic cytokine, serving as a primary driver of renal fibrosis in DKD [192,193]. DKD is linked to various detrimental factors, including elevated glucose levels, AGEs, hypertension, and dyslipidemia. These factors can trigger TGF-β signaling through both TGF-β-dependent and independent pathways [193]. High glucose levels amplify TGF-β signaling and boost the activity of specific elements involved in fibronectin transcription in various types of kidney cells, including mesangial, fibroblast, and proximal tubular cells [193,194]. This influence on TGF-β expression might be linked to a glucose-responsive element in the Tgfb1 gene’s promoter [195]. Additionally, high glucose levels enhance the activity of TGF-β1 by increasing thrombospondin 1 (TSP1), which can activate latent TGF-βs [196,197]. Moreover, high glucose independently elevates the transcription of TGF-β receptor II in murine mesangial cells, even without the induction of TGF-β [198]. Thus, high glucose can activate TGF-β signaling during the development of DKD.
In both patients and animal models with DKD, there is a substantial increase in the expression or activation of TGF-β ligands, TGF-β receptors, and the subsequent signaling mediators like Smad2 and Smad3 in glomeruli, tubules, and the tubulointerstitium [46,198,199,200,201]. This garnered substantial attention in the field, as evidenced by seminal studies demonstrating the protective effects of TGF-β antibodies [202]. TGF-β, operating through both Smad-dependent and -independent signaling pathways, assumes a pivotal role in myofibroblast activation. This leads to heightened production of ECMs and suppression of ECM turnover or degradation [203]. Ultimately, this results in an excessive buildup of ECM, a prominent hallmark of renal fibrosis. Moreover, it is evident that TGF-β also plays a role in inducing EMT, as described earlier, as well as endothelial to mesenchymal transition. These processes involve the conversion of local resident cells into those with more pro-sclerotic traits, thereby contributing to kidney injury and fibrosis [204].
The success of inhibiting TGF-β1 signaling in animal studies has prompted clinical investigations in DKD [205,206]. While pirfenidone, an antifibrotic agent, increased eGFR in a diabetic patient cohort [205], a placebo-controlled phase II study utilizing a humanized TGF-β1-specific neutralizing monoclonal antibody alongside renin–angiotensin system blockades did not succeed in slowing the progression of DKD in patients [206].
5.2. MAPK Signaling Pathway
MAPKs, a set of serine/threonine protein kinases, oversee a range of cellular functions and can become active under high-glucose conditions [191,207]. The primary subgroups within the MAPK family are P38/MAPK, ERK, and JNK, collectively playing a role in signal transduction during fibrosis [191]. Various studies have shown that modulating specific components of the MAPK signaling pathways can have significant effects on renal fibrosis in DKD. Increasing EphA1 expression was found to reduce phosphorylation of ERK1/2 and JNK, leading to the alleviation of renal fibrosis in mice models of DKD [208]. Additionally, the ERK1/2 MAPK pathway was identified as a crucial player in DKD, influencing fibrogenesis by regulating mesangial cell activities and ECM accumulation [209]. Inhibition of p38 MAPK showed promise in reducing the presence of phosphorylated p38 MAPK-positive cells, particularly in the early stages of fibrosis [210]. Targeting p38 MAPK exclusively was effective in alleviating renal fibrosis in an established fibrotic model [211]. Moreover, inhibiting JNK isoforms significantly delayed fibrosis progression by impeding the accumulation of collagen IV and α-SMA+ myofibroblasts in an animal model [212]. These findings collectively highlight the potential therapeutic targets within the MAPK pathways for managing renal fibrosis in DKD [191]. In an in vitro study, it was demonstrated that the activation of p38 MAPK and ERK in human podocyte cells through IL-17RA [134] while another study showed that pentosan polysulfate mitigated apoptosis and inflammation by suppressing the activation of the p38 MAPK pathway in human renal proximal tubular epithelial cells (HK 2) treated with high glucose [213]
5.3. Wnt/β-Catenin Pathway
The Wnt signaling pathway can be categorized into two primary pathways: the canonical β-catenin-dependent pathway and the non-canonical β-catenin-independent pathway [214]. The most extensively studied Wnt signaling pathway is the canonical Wnt/β-catenin pathway Recent studies proved that dysregulation of the Wnt signaling pathways participates in the occurrence and progression of type 2 DM by directly influencing the differentiation and proliferation of pancreatic β-cells and the secretion and action of insulin [215]. During CKD, the Wnt canonical signaling pathway becomes active, whereas it remains comparatively inactive in the normal adult kidney [216,217]. The Wnt canonical and non-canonical pathways have been identified as another key player in the progression of DKD [218,219]. Dysregulated Wnt signaling, which causes harm to podocytes [220,221,222] and mesangial cells [223,224,225], ultimately culminates in renal fibrosis [214]. An earlier study established that aberrant upregulation of the Wnt/β-catenin signaling pathway leads to podocyte damage and dysfunction [220]. The study reported that abnormal activation of the Wnt/β-catenin pathway hinders WT1 (a podocyte marker)-mediated gene expression, resulting in podocyte de-differentiation and mesenchymal transformation [221]. Conversely, blocking the Wnt/β-catenin pathway reinstates WT1-mediated gene regulation, thus preserving podocyte integrity [221]. Likewise, DM induces the production of Ras/Rac1-dependent superoxide, subsequently impeding Wnt proteins in mesangial cells [226]. In a murine model of DM, treatment with superoxide dismutase-conjugated propylene ethyl glycol demonstrated an improvement in the detrimental effects of free radicals on mesangial cells. High glucose initiates the activation of GSK-3β, resulting in the instability and degradation of β-catenin. This, in turn, induces mesangial cell apoptosis by promoting the cleavage of caspase-3 and polyADP-ribose polymerase (PARP) [224]. A previous study showed that transfecting mesangial cells with WNT4, WNT5a, or stable β-catenin (S33Y) can hinder the activation of GSK-3β and enhance the stability of nuclear β-catenin, thereby reducing the level of apoptosis in mesangial cells [225]. The use of medications such as simvastatin [227] and spironolactone [228] leads to increased secretion of Wnt5a protein in mesangial cells in patients with DKD. This heightened secretion of Wnt5a facilitates the translocation of β-catenin to the nucleus, thereby enhancing the protective effect against high glucose-induced inhibition of Wnt/β-catenin. This process ultimately safeguards glomerular mesangial cells and contributes to a beneficial outcome for individuals with DKD. Hence, examining the expression of Wnt proteins holds significant relevance for DKD.
5.4. PI3K/AKT, JAK/STAT, and Notch Pathways
The PI3K/AKT signaling pathway assumes a central regulatory role in the progression of DKD. In DM, this pathway becomes activated in renal tubular cells, overseeing processes such as cell growth, EMT, and lipid metabolism [229,230]. Using a murine model of DKD, a recent study indicates that METTL14 (a core component of methyltransferase complex)-regulated PI3K/AKT signaling pathway via PTEN affected histone deacetylase 5 (HDAC5)-mediated EMT of renal tubular cells [231]. The PI3K/AKT/GSK-3β pathway can mediate oxidative stress and apoptosis in DKD. Using a mouse model of STZ-induced DKD, Wang et al. have demonstrated that phillyrin (a major active component of Forsythia suspensa) inhibited glycogen synthase kinase-3β (GSK-3β) activity by activating the PI3K/AKT signaling pathway, increased the Bcl-2/Bax ratio, reduced the release of cytochrome c from the mitochondria to the cytoplasm, subsequently inhibited the activation of caspase-3 and ultimately suppressed renal cell apoptosis [232]. The PI3K/AKT signaling pathway, in DKD, was also shown to regulate oxidative stress and inflammation by controlling GSK-3β/Nrf2 and ASK1/JNK signaling pathways, respectively [233]. Increasing evidence suggests that many signaling pathways in DKD have been implicated in AKT phosphorylation, and activation of AKT is required for DKD occurrence and development [234,235,236]. AKT, in conjunction with glycogen synthase kinase-3β (GSK-3β) as its downstream mediator, is instrumental in regulating mitochondrial functions [237]. Within a high glucose environment, mitochondrial dysfunction has a pivotal role in both the onset of oxidative burst and the initiation of apoptosis [238]. Recent studies have underscored the interrelation between AKT activation and mitochondrial function, oxidative stress, and apoptosis [239,240]. The phosphorylated form of AKT elicits an opposing regulatory influence on GSK-3β [236]. In the context of DKD, AKT phosphorylation is restrained, leading to the activation of GSK-3β [233]. Subsequently, this activated GSK-3β orchestrates the equilibrium between Bax and Bcl-2, thereby influencing mitochondrial permeability. This instigates the opening of the mitochondrial permeability transition pore, culminating in the liberation of cytochrome C from the mitochondria, ultimately contributing to the regulation of cell apoptosis [241,242].
Recent studies focused on JAK/STAT signaling shed light on its involvement in various factors linked to the progression of DKD including fibrosis, immunity, inflammation, aging, autophagy, and EMT. Thus it stands as the central signaling hub in the advancement of DKD [243]. A direct relationship between tubulointerstitial JAK/STAT expression and progression of kidney failure in patients with DKD has been observed [244]. The activation of JAK/STAT signaling pathways has been implicated in tubulointerstitial fibrosis and epithelial to mesenchymal transition in various conditions, including DM, as demonstrated in animal models [245,246,247]. Additionally, JAK/STAT activation has been observed in rat glomerular cells exposed to high glucose [248,249]. In the STZ-induced DKD mouse, findings illustrate that elevated glucose levels lead to the activation of JAK2 and the STATs, primarily through an AngII-dependent mechanism leading to the initial kidney damage [248]. Furthermore, elevated glucose levels stimulate the proliferation of glomerular mesangial cells (GMC) and trigger the production of TGF-β. Concurrent incubation with AG-490, a specific JAK2 inhibitor, effectively counteracted the high glucose-induced surge in TGF-β and fibronectin synthesis. AG-490 also nullified the tyrosine phosphorylation of JAK2, STAT1, and STAT3 induced by high glucose in GMC. When GMC cultured in 25 mmol/L glucose were preincubated with a specific JAK2 or STAT1 antisense oligonucleotide, both TGF-β and fibronectin synthesis were prevented. These findings offer direct evidence of the connections between JAK2, STAT1, and the excessive production of TGF-β and fibronectin induced by glucose in GMC [249]. Moreover, in the study by Zheng et al., it was discovered that suppressing STAT3 resulted in an enhancement of renal function and a reduction in the expression of TGF-β1, VEGF, and other factors associated with fibrosis and collagen buildup in C57BL/6 diabetic mice [250].
The Notch signaling pathway in the mammalian system comprises four transmembrane receptors (Notch1–Notch4), two Jagged family ligands (JAG1 and JAG2), and three delta-like ligands (DLL1, DLL3, and DLL4) [191,251]. Activation of the Notch signaling pathway was observed in patients with tubular interstitial fibrosis (TIF) as well as in TIF mouse models. Additionally, it was established that the Notch pathway is both necessary and sufficient for the initiation and progression of TIF [191]. In a mouse model of DKD, it was demonstrated that gliquidone, a diabetic medication, can ameliorate the diabetic symptoms of DN by inhibiting the Notch/Snail1 signaling pathway, improving anti-oxidative response, and delaying renal interstitial fibrosis in a dose-dependent manner [252]. In another study by Nishad et al., both in vitro using immortalized human podocytes and in a mouse model, it was shown that excess growth hormone (GH) activates Notch1 signaling in a γ-secretase-dependent manner. Pharmacological inhibition of Notch1 using the γ-secretase inhibitor DAPT (N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-S-phenyl glycine t-butylester) resulted in reduced podocyte loss. Importantly, the results demonstrated that DAPT treatment halted cytokine release and prevented glomerular fibrosis, all of which were induced by excess GH. Furthermore, DAPT prevented GH-induced GBM thickening and proteinuria [253]. Moreover, the Notch signaling pathway may regulate oxidative damage and apoptosis in glucose-mediated renal tubular epithelial cells by controlling mitochondrial dynein and biogenesis genes, potentially accelerating renal interstitial fibrosis in DKD [254].
5.5. Mineralocorticoid Receptor (MR) and Aldosterone
Mineralocorticoids are a class of steroid hormones, with aldosterone being the primary physiological mineralocorticoid. It is produced in the outer layer of the adrenal gland as a response to hyponatremia and hyperkalemia, triggered by the activation of the RAAS [255]. There is growing evidence indicating the existence of an intricate network involving aldosterone, the MR, and Ras-related C3 botulinum toxin substrate 1 (Rac1) as crucial elements in the generation of ROS and subsequent damage caused by oxidative stress. This dynamic interaction plays a significant role in initiating interstitial nephritis, ultimately culminating in fibrosis in cases of DKD [256].
The MR functions as intracellular receptors, operating as a nuclear transcription factor or exerting rapid non-genomic effects through secondary cell signaling pathways [257,258]. The MR was initially believed to exclusively bind with mineralocorticoids, but subsequent research revealed that aldosterone, cortisol, and progesterone bind to the MR with equal affinity [259]. The MR instigates the inflammatory cascade by generating ROS through NADPH in the mitochondria, a process further amplified by Rac1 [260]. Additionally, aldosterone augments the expression and activation of serum- and glucocorticoid-inducible protein kinase 1 (SGK1), a factor associated with the development of renal fibrosis [260]. In a study using uninephrectomy in type 2 diabetic mouse models, it was It was demonstrated that salt-induced activation of the Rac1-MR pathway in distal tubules and glomeruli played a role in DKD progression through hypertension and glomerular injury, respectively. This discovery underscores the potential of MR antagonism in conjunction with Rac1 inhibition as a novel strategy for DKD treatment [261]. Both Rac1 and aldosterone contribute to fibrosis by activating inflammasomes. Studies indicate that exposing macrophages to elevated levels of aldosterone leads to inflammasome activation via mitochondria-derived ROS. This inflammasome activation in macrophages has been shown to mediate aldosterone-induced renal fibrosis in a murine model [262]. The expression of a well-studied inflammasome, NLRP3, was shown to be heightened in the glomeruli of DKD patients, correlating with the degree of albuminuria [263]. Within podocytes, aldosterone, acting through an MR, in conjunction with Rac1, triggers the activation of the NLRP3 inflammasome, resulting in podocyte injury and glomerular sclerosis [264].
Aldosterone triggers the activation of inflammasomes in macrophages, provoking an inflammatory tubulointerstitial region [262]. Additionally, macrophages play a crucial role in generating renal TGF-β, a key factor in renal fibrosis [265]. The pathway leading to TGF-β-driven renal fibrosis seems to be facilitated by Rac1, which acts as a substantial redox-dependent non-SMAD (noncanonical) regulatory factor [265]. The role of the MR and aldosterone is depicted in Figure 6.
While it is acknowledged that aldosterone exhibits MR-independent effects contributing to DKD development, and Rac1 can be upregulated without MR activation, MR remains a pivotal component in this interaction. Clinical evidence supports the efficacy of the novel non-steroidal MR antagonist (nsMRA), finerenone, in slowing the progression of DKD [256].
5.6. OPN-Mediated Fibrosis in DKD
OPN is a secreted, pleiotropic, multi-phosphorylated glycoprotein, first recognized as secreted phosphoprotein 1 (SPP1) in 1979 [266]. The kidneys possess the highest concentration of OPN compared to other tissues [267]. During kidney injury, its presence is significantly increased in all tubular segments and especially in the glomeruli [268]. The role of OPN in DKD has been reviewed in detail by our group recently [269]. During the last decade, a number of studies analyzed the role of OPN in the pathogenesis of DKD and reported high expression of OPN in the tubular epithelium of the renal cortex and in glomeruli in rat and mouse models of DKD [270,271]. Our lab has shown that TGF-β likely mediates the effect of OPN in DKD mouse models enhancing glomerular damage [14]. In this study, we generated STZ-induced experimental, and genetic models of type 1 (Ins2Akita) and type 2 (Leprdb/db) diabetic mice on the background of OPN-null and wild-type mice. In both mouse models, OPN deletion decreased albuminuria, glomerular mesangial area, fractional volume of expansion, and expression of glomerular collagen IV, fibronectin, and TGF-β in the diabetic mice compared with their respective controls. In in vitro experiments with cultured mouse mesangial cells, the TZDs, rosiglitazone and pioglitazone, but not insulin, decreased AngII-induced OPN expression, while recombinant OPN upregulated TGF-β, ERK/MAPK, and JNK/MAPK signaling, which have been shown to be involved in the pathology of DKD. These studies strongly suggested that OPN expression enhanced glomerular damage, likely through the expression of TGF-β [14]. Our findings gained further support from a recent investigation in the German Chronic Kidney Disease cohort [272]. This study revealed a correlation between elevated OPN levels and a deterioration in kidney function markers, along with an increased likelihood of experiencing adverse outcomes. The researchers concluded that a significant portion of kidney function decline could be attributed to heightened OPN levels [272]. The full-length OPN protein is cleaved by various proteases, including thrombin, matrix metalloproteinase (MMP)-3, MMP-7, cathepsin-D, and plasmin, producing ntOPN, which may have more detrimental effects in CKD [269]. Therefore, we further investigated whether ntOPN may be a better predictor of DKD, and this hypothesis was subsequently tested in a Chinese population [15]. Collectively, our research showed that urinary ntOPN stands out as an independent marker for both the initiation and the progression of DKD. When compared to conventional biomarkers like serum creatinine combined with urinary albumin-to-creatinine ratio, our multi-biomarker models centered around urinary ntOPN, demonstrated enhanced predictive power in forecasting DKD progression. This development holds the potential to provide a more accurate biomarker for evaluating the risk of DKD in individuals with DM [15]. Another study, although in a different scenario, demonstrated that milk ntOPN bounded intestinal cells most effectively and was transported across the intestinal membrane indicating that proteolytic processing of OPN to form ntOPN increases its biological activity [273]. These findings support the efficacy of OPN/ntOPN as a therapeutic target in DKD. The OPN-mediated signaling pathways are shown in Figure 7.
6. Genetics and Epigenetics of DKD
Studies have demonstrated a familial clustering of DKD across diverse ethnic groups, suggesting a genetic contribution to its development. Additionally, genetic risk factors in DKD interact with environmental elements such as lifestyle, diet, and medication. The interplay between genetic, epigenetic, and environmental factors in the initiation and progression of DKD has been illustrated in several studies [274,275,276,277,278]. Genetic investigations of DKD primarily focus on analyzing associations between variations in genomic DNA, such as single nucleotide polymorphisms (SNPs), copy number variants (CNVs), and microsatellites, with the clinical manifestations of the disease [274,276,279,280]. Notably, significant linkage peaks have been observed on chromosomes 3q and 18q, implicating genes like AngII receptor type 1 [281], adiponectin [282], non-catalytic region of tyrosine kinase adaptor protein 1 (NCK1) [283], and carnosine dipeptidase 1 (CNDP1) [284] in DKD susceptibility. Recently, a meta-analysis of genome-wide association studies (GWAS), including 19,406 individuals of European ancestry with type 1 DM was performed by Salem et al. Within this analysis, they found 16 risk loci of genome-wide significance. Notably, the most strongly associated variant (rs55703767) is a prevalent missense mutation found in the collagen type IV alpha 3 chain (COL4A3) gene that encodes a vital structural element of the glomerular basement membrane [285]. In a separate investigation involving 13,123 individuals with type 2 DM, a meta-analysis uncovered a fresh link between DKD and the variant rs72763500 (located at chr1:236116561). This variant serves as a splicing quantitative trait locus for the nidogen-1 (NID1) gene. NID1, a significant constituent of the basement membrane in renal tubules, potentially exerts an influence on the development of DKD in T2D [286]. Further, in a thorough GWAS study mega-analysis encompassing 33,879 patients, scientists pinpointed particular SNPs (specifically, rs3128852, rs117744700, and rs28366355) linked to DKD. Importantly, they confirmed the causative connection between rs3128852 and the onset of DKD [287]. Through GWAS of the Family Investigation of Nephropathy and Diabetes cohort, we have identified several novel loci associated with albuminuria, eGFR, DM, DKD, advanced DKD, and renal failure [288,289,290,291,292,293]. This discovery not only elucidated the genetic landscape of DKD but also paved the way for further exploration through functional analyses. Additionally, it offered new prospects for developing innovative diagnostic markers for DKD. Hence, these studies represented progress toward a broader comprehension of the underlying mechanisms of DKD.
On the other hand, in the realm of DKD, epigenetic investigations focus on potentially heritable changes in gene expression that arise without any alterations in the underlying DNA sequence [277,294,295]. These studies reveal how environmental factors can influence the patterns of gene expression implicated in the progression of DKD. For instance, a comprehensive analysis of methylomes, transcriptomes, and genetic variations in 500 patients with DKD identified 40 loci displaying altered methylation and expression strongly tied to DKD. These loci demonstrated functional connections to processes like complement activation, inflammation, and apoptosis [296]. Another study identified 19 CpG sites linked to eGFR, including five associated with confirmed renal fibrosis, indicating DNA methylation alterations in the kidney cortex [297,298]. Additionally, abnormal DNA methylation patterns were noted in proximal tubular epithelial cells from the murine model of DM, with genes functionally associated with mitochondrial biogenesis [299]. The interplay between gene methylation levels and clinical markers of DKD is significant [300,301]. For example, elevated albuminuria correlated with hypomethylation of tissue inhibitor metalloproteinase-2 (TIMP-2) and aldo-keto reductase family 1 member B [302], while higher serum homocysteine levels were observed in DKD patients with increased promoter methylation of the methylenetetrahydrofolate reductase gene [303]. CTGF, associated with extracellular matrix accumulation and DKD pathogenesis, exhibited lower DNA methylation levels and increased protein levels, correlating with albuminuria and eGFR decline in DKD patients [304]. Furthermore, the RAS protein activator like 1 (Rasal1) gene, displayed hypermethylation of its promoter in DKD, contributing to fibroblast activation and kidney fibrosis progression [305]. Changes in Rasal1 promoter DNA methylation were reversed by tet methylcytosine dioxygenase 3-mediated hydroxymethylation, with a concomitant reduction in fibrosis [305]. In summary, these findings emphasize the dynamic regulation of multiple signaling pathways by DNA methylation in the progression of DKD.
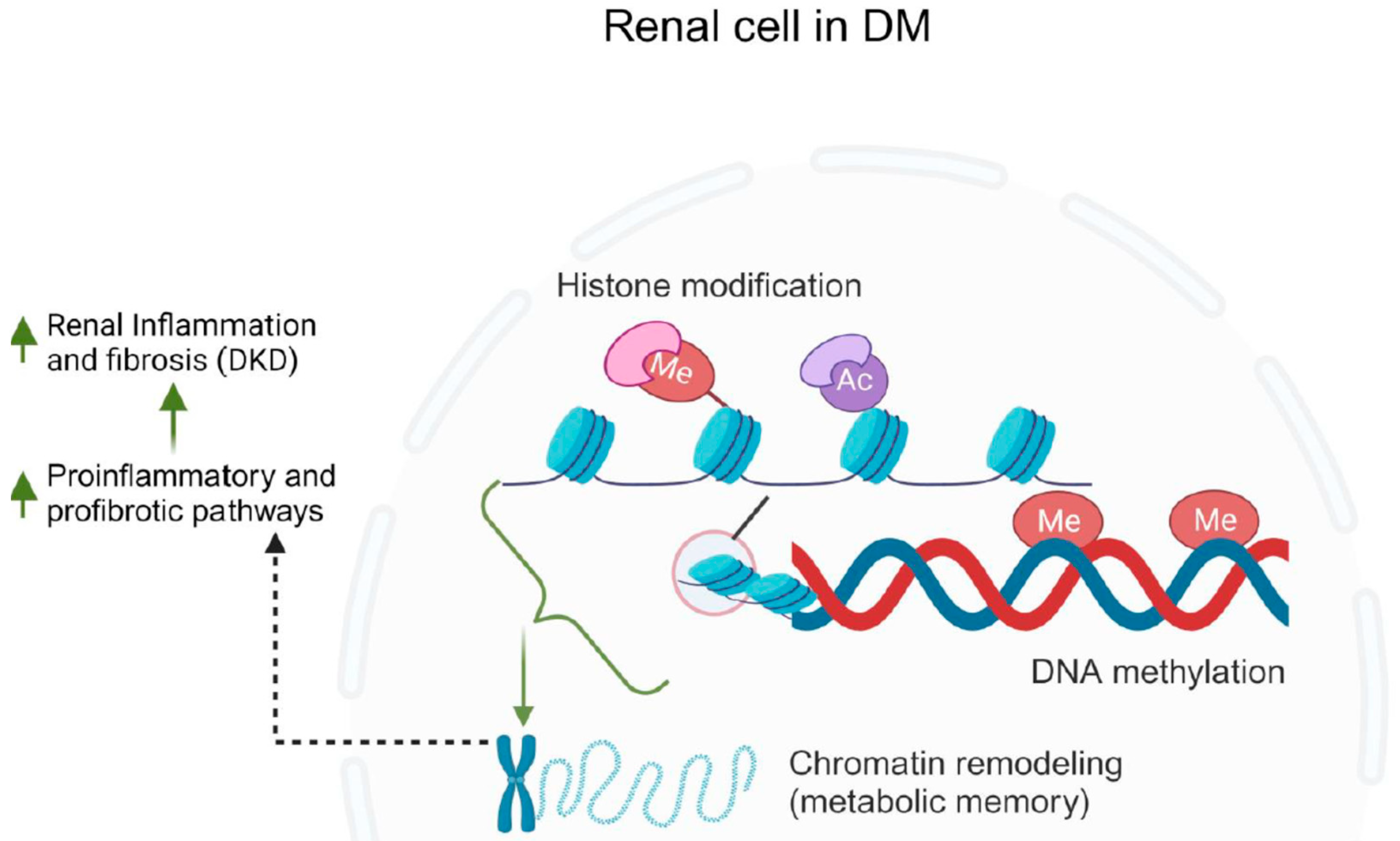
In the context of DKD, histone modifications are crucial in regulating gene expression patterns that contribute to the progression of the disease [306,307]. For instance, studies have shown that certain histone modifications, such as acetylation and methylation, are associated with the dysregulation of genes involved in inflammation [308,309,310,311], fibrosis [312,313], and oxidative stress responses [314,315] in DKD. These modifications create a molecular environment that promotes the activation of pro-inflammatory and pro-fibrotic pathways, ultimately leading to the structural and functional changes observed in diabetic kidneys. Histone methylation occurs at arginine, lysine, and histidine sites located on histone tails [316] that can either enhance or inhibit gene transcription [311]. Lysine methylation is regulated by enzymes known as methyltransferases (KMTs) and can be reversed by demethylases (KDMs) [317,318,319]. One category of KMTs includes enzymes containing SET domains [317]. In diabetic mice, there was an increase in KDM6A expression and a decrease in H3K27me2/3 levels observed in the kidneys [320]. This pattern was also evident in podocytes and renal tubular cells subjected to hyperglycemic conditions [321,322]. Moreover, kidney tissues from individuals with DKD displayed reduced levels of H3K27me3 in podocytes, glomerular cells, and tubular cells [323]. Comparatively, diabetic animals exhibited elevated levels of H3 lysine 4 mono-methylation (H3K4me1) and H3K4me3, along with decreased levels of H3K27me3 in the kidneys when compared to non-diabetic controls [324]. Moreover, being exposed to high blood sugar levels early in life, particularly during fetal development, heightens the risk of developing type 2 DM in adulthood [325]. There is mounting evidence suggesting that prolonged hyperglycemia induces a “metabolic memory” through epigenetic alterations in DKD, which alter gene expression [311,325]. The role of epigenetics in DKD is summarized in Figure 8.
7. Biomarkers Associated with DKD
In DKD, biomarkers offer valuable insights into the early detection, progression, and management of the condition, enabling timely interventions to slow or halt its progression. Additionally, they facilitate research efforts aimed at understanding the underlying mechanisms of DKD and developing innovative therapies. Overall, biomarkers serve as indispensable tools in the fight against DKD, contributing to improved patient outcomes and quality of life. There are several important DKD biomarkers that have been studied, as shown below in Table 1.
7.1. Biomarkers Associated with Tubular Damage in DKD
Several biomarkers for tubular damage in DKD have been identified. For example, plasma kidney injury molecule-1 (KIM-1) is a notable indicator of early tubular injury and stress in DKD [176,326,327]. Markers like TNFR-1 and TNFR-2, receptors for tumor necrosis factor [176,326,328], along with neutrophil gelatinase-associated lipocalin (NGAL) [327,329,330] provide insights into acute kidney injury and inflammation, aiding in the evaluation of tubular damage. The urinary liver-type fatty acid-binding protein (L-FABP) is specific to proximal tubules and its presence in urine indicates early tubular damage [331,332]. OPN [14] and ntOPN [15], a protein associated with kidney injury, contribute to understanding tubular damage in DKD. Urinary AngII-converting enzyme 2 (ACE2), a component of the renin–angiotensin system, and angiotensin, known for their roles in blood pressure regulation, also offer valuable indicators of tubular health [333,334]. Additionally, markers such as n-acetyl-β-D-glucosaminidase (NAG) [334,335], α1-microglobulin [336,337], fibroblast growth factor 23 (FGF-23) [338], α-klotho [339], and a disintegrin and metalloprotease-10 (ADAM-10) [340] play pivotal roles in assessing tubular function and damage. This comprehensive array of biomarkers provides a multifaceted approach to evaluating tubular health and damage in individuals with DKD.
7.2. Biomarkers Associated with Glomerular Damage in DKD
Similarly, biomarkers of glomerular damage play a critical role in evaluating DKD. Transferrin, a glycoprotein, has shown promise as a potential biomarker, as elevated levels have been associated with glomerular dysfunction in DKD [341,342,343]. Type IV collagen, a structural protein in the basement membrane of glomeruli, is another key biomarker [344,345]. Elevated levels of type IV collagen indicate basement membrane thickening and glomerular damage in DKD. Cystatin C, a protein freely filtered by the glomerulus, offers insights into the glomerular filtration rate and serves as an indicator of glomerular health [346,347]. Ceruloplasmin, an enzyme involved in iron metabolism, has also been implicated in DKD and may serve as a biomarker for glomerular damage [342]. Fibronectin, an adhesive glycoprotein, is associated with glomerular injury and fibrosis [348]. Urinary podocytes–podocalyxin [349], and vascular endothelial growth factor (VEGF) [337] predict the progression of the disease and are shown to significantly correlate with urinary albumin excretion. Additionally, α-klotho [350], and ADAM-10 [340,351] play crucial roles in glomerular health and function. Also, urinary proteomics identifies cathepsin D as a biomarker of rapid eGFR decline in type 1 diabetes [352]. Together, these biomarkers provide a comprehensive toolkit for assessing glomerular health and damage in individuals with DKD.
Recently, urine levels of adenine, a metabolite produced in the kidney, was shown to be predictive and a causative biomarker of looming progressive kidney failure in patients with DM, a finding that could potentially lead to earlier diagnosis and intervention. The elevated adenine was also associated with all-cause mortality [353]. This was the first study to also show that an inhibitor of endogenous adenine was nephroprotective.
7.3. Inflammatory and Oxidative Stress Biomarkers of DKD
Inflammatory and oxidative biomarkers are additionally pivotal in DKD development. Elevated markers like TNF-α signify progression to severe stages, including ESRD and GFR loss [354]. MCP-1 predicts renal disease progression, correlating with albuminuria and exacerbating inflammation and fibrosis [355]. TGF-β is linked to higher mortality risk and macroalbuminuria [193] while interleukins (IL-1β, IL-6, IL-8, IL-18) predict early renal decline risk [356]. These biomarkers highlight chronic inflammation in DKD. Likewise, oxidative stress, indicated by markers like 8-hydroxydeoxyguanosine (8oHdG) [357], pentosidine [358], uric acid [359], malondialdehyde (MDA) [360], superoxide dismutase (SOD) [360], and glutathione peroxidase (GPx) [361], contributes significantly to DKD. The interplay of inflammation and oxidative stress worsens kidney injury. Understanding and monitoring these biomarkers offer crucial insights into DKD mechanisms, suggesting potential therapeutic targets.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Renal biomarkers associated with DKD.
| Tubular | Glomerular | Inflammatory/Oxidative Stress |
|---|---|---|
| KIM-1, TNFR-1, and TNFR-2 [176,326,327,328] | Transferrin [341,342,343] | TNF-α [354] |
| NGAL [327,330] | Type IV collagen [344,345] | MCP-1 [355] |
| Urinary L-FABP [331,332] | Cystatin C [346,347] | TGF-beta [193] |
| OPN [14], ntOPN [15] | Ceruloplasmin [342] | ILs [356] |
| ACE2 [333,334] | Fibronectin [348] | 8oHdG [357] |
| NAG [334,362] | Podocytes-podocalyxin [349] | Pentosidine [358] |
| α1-microglobulin [336,337] | VEGF [337] | Uric acid [359] |
| FGF-23 [338] | α-alpha-Klotho [350] | MDA [360] |
| α-Klotho [339] | ADAM-10 [340,351] | SOD [360] |
| ADAM-10 [340] | Cathepsins [352] | GPx [361] |
| Adenine [353] | Adenine [353] |
Abbreviations: KIM-1: kidney injury molecule-1, TNFR: TNF-α receptor, NGAL: neutrophil gelatinase-associated lipocalin, L-FABP: liver-type fatty acid-binding protein, OPN: osteopontin, ntOPN: N-terminal OPN, ACE2: angiotensin-converting enzyme 2, NAG: N-acetyl-β-D-glucosaminidase, FGF-23: fibroblast growth factor 23, ADAM-10: a disintegrin and metalloprotease 10, VEGF: vascular endothelial growth factor, MCP-1: monocyte chemoattractant protein-1, TGF-β: transforming growth factor-β, ILs: interleukins, 8-Ohdg: 8-hydroxydeoxyguanosine, MDA: malondialdehyde, SOD: superoxide dismutase, GPx: glutathione peroxidase.
8. Therapies Targeting Specific DKD Pathomechanisms
Continuous understanding of the mechanisms involved in the development of DKD has led to the development of several pathomechanism-targeted therapies over the past two decades. For instance, until recently, the mainstay of therapy for DKD highlighted metabolic and hemodynamic-related therapies via optimization of glucose and blood pressure control and the use of RAS inhibitors, namely ACE inhibitors and ARBs. Despite these measures, significant residual risk for DKD progression remained, which then led to the discovery of newer therapies that have emerged as both reno- and cardio-protective agents. In particular, SGLT2 inhibitors, function as another hemodynamic target, and nsMRAs, interrupt aldosterone signaling via the mineralocorticoid receptor, which undergoes over activation in DKD, have provided significant hope and continue to extend kidney life and the overall lifespan for patients with DKD. A summary of the available treatment options in DKD is presented in Table 2.
8.1. RAAS Inhibitors
The Kidney Disease Improving Global Outcomes (KDIGO) as well as the American Diabetes Association (ADA) guidelines have recommended the use of ACE inhibitors and ARBs for patients with DKD to reduce blood pressure and to achieve ≥30% reduction in albuminuria [363,364,365]. In addition to their hemodynamic effects, described above, ACE inhibitors and ARBs can block oxidative stress and pro-inflammatory pathways. Despite their renal benefit to reduce DKD progression observed from large kidney outcome trials, Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan [125] and the Irbesartan Diabetic Nephropathy Trial [136], ACE inhibitors and ARBs have demonstrated no evidence to reduce the composite of cardiovascular endpoints (cardiovascular death, myocardial infarction, hospitalization for heart failure, hospitalization for angina or lower limb amputation above the ankle).
8.2. SGLT2 Inhibitors
The individual KDIGO and ADA guidelines as well as the KDIGO-ADA consensus report have recommended the use of SGLT2 inhibitors in patients with DKD and cardiovascular disease on top of standard of care to address the residual risk of DKD progression. These new therapies function through the reduction of tubuloglomerular feedback and glomerular hyperfiltration, as we described above [363,364,365]. Notably, the broad renal and cardiovascular benefits of this new class of drugs may not be fully explained through this mechanism. Emerging evidence suggests that SGLT2 inhibitors may function via other mechanisms including blockade of the SGLT2 nutrient surplus sensor capacity [366,367] as well as other pleiotropic mechanisms [368]. For example, the inhibition of SGLT2 acts as a protective measure against organ hypoxia, a pathway frequently linked to the progression of DKD due to its role in fostering and sustaining fibrotic and inflammatory responses [369,370]. Additionally, studies on human proximal tubular cells, murine models of type 2 DM, and patients with type 2 DM have revealed that the inhibition of systemic SGLT2 results in a reduction of various molecules associated with inflammation, extracellular matrix turnover, and fibrosis. These molecules include TNFR1, MMP-7, IL-6, and fibronectin 1 [371,372,373,374]. A large meta-analysis of placebo-controlled trials indicates that the use of SGLT2 inhibitors modifies the risk of CKD progression and acute kidney injury, not only in patients with type 2 DM at high cardiovascular risk but also in patients with CKD or heart failure irrespective of DM [375].
8.3. nsMRAs
The nsMRAs have emerged as novel anti-inflammatory and anti-fibrotic therapies with proven efficacy to reduce the risk of renal function decline as well as ESRD, cardiovascular death, non-fatal heart attacks, and hospitalization for heart failure. While finerenone is the first-in-class nsMRA approved for clinical use, there are several other selective nsMRAs with anti-inflammatory and anti-fibrotic properties undergoing assessment through clinical trials [376,377].
8.4. ET Receptor Antagonist
ET, a polypeptide, is known as a potent vasoconstrictor [378,379]. It is produced by epithelial and mesangial cells in the kidney and plays a crucial role in regulating blood flow, and glomerular filtration, as well as water, sodium, and acid-base balances [379,380,381]. ET exerts its effects through ET receptors A (ETA) and B (ETB), expressed on various renal components including glomerular podocytes, mesangial cells, and arterioles [379,380,381]. ET’s activation of these receptors can have detrimental effects on the kidney, contributing to the progression of DKD [382,383,384]. Therefore, blocking ET receptors with ET receptor antagonists (ERAs) like Atrasentan, has been shown to have protective effects on the kidneys in patients with DM [385]. ERAs impact glomerular hemodynamics, leading to improved blood pressure, reduced proteinuria, and a balanced filtration rate [382,386]. They also provide kidney protection by addressing issues like podocyte injury [382,387], reducing mesangial matrix accumulation [388], and mitigating fibrosis and inflammation [389,390]. This leads to a decrease in glomerular permeability and proteinuria [391]. Nonetheless, findings from clinical trials suggest that these substances can also lead to side effects related to fluid volume [391,392]. Combining ERAs with SGLT2 inhibitors or utilizing dual Agn-II type 1/ET receptor blockers have been suggested as strategies to overcome these challenges [379,393].
Table 2.
Summary of the available medications to treat patients with DKD.
| Targeted Pathways | Therapeutic Function | Clinical Outcomes | References |
|---|---|---|---|
| RAAS Blockers | Reducing inflammatory and fibrotic processes | Slow the deterioration of kidney function. Reduce the risk for creatinine doubling, and ESRD. | [125,136,394,395] |
| SGLT2 inhibitors | Blocking reabsorption of glucose in the proximal tubule | Minimize significant eGFR decline, kidney failure, heart failure, and mortality due to kidney and CVD. | [372,396,397,398,399] |
| Nonsteroidal mineralocorticoid receptors (MRs) antagonist | Downregulating inflammatory and fibrotic pathways in nonepithelial cells; Downregulating ion channels for sodium and potassium in kidney tubular epithelial cells | Minimizes significant eGFR decline, kidney failure, heart failure, and mortality due to CKD. | [400,401,402] |
| Endothelin receptor antagonist | Downregulating inflammatory and fibrotic pathways; Efferent artery vasodilation | Reduces albuminuria, eGFR decline, kidney failure as well as death from CKD. | [385,403] |
Abbreviations: RAAS: renin–angiotensin–aldosterone system, ESRD: end stage renal disease, SGLT2: sodium–glucose cotransporter 2, eGFR: estimated glomerular filtration rate, CVD: cardiovascular disease, MR: mineralocorticoid receptor CKD: chronic kidney disease.
9. Conclusions and Future Directions
There remains a notable gap in our understanding of DKD that warrants further research. While significant progress has been made in identifying key factors and biomarkers, understanding the underlying molecular mechanisms, and developing targeted therapies, there is still much to uncover. One area of interest is the intricate interplay between genetic predisposition and environmental factors in the development and progression of DKD. The future of managing DKD encompasses a comprehensive approach. This involves tailoring therapies based on an individual’s genetic, molecular, and clinical profile. In DKD, maintaining precise glycemic control is essential, coupled with innovative approaches targeting inflammation and fibrosis. The modulation of the RAAS pathway holds particular significance. Additionally, the exploration of medications like SGLT2 inhibitors, and nsMRAs, as well as GLP-1 receptor agonists have shown considerable promise. Furthermore, exploring the potential benefits of combination therapies, such as the use of ERAs in conjunction with SGLT2 inhibitors, holds potential for improving outcomes in DKD patients. Moreover, long-term studies assessing the efficacy and safety of emerging treatments are essential to establish their viability in clinical practice. As research continues, new knowledge is gained, increasing the likelihood of identifying innovative targets that may potentially and completely eliminate the risk of CKD progression.
Early biomarker discovery and advances in regenerative medicine are emerging focal points in this field. Notably, our lab is actively researching ntOPN as a potential early biomarker and therapeutic target with the capacity to significantly reduce fibrosis and inflammation in DKD. In tandem with these pharmacological strategies, integrated care, and patient education are pivotal. Encouraging diversity in the participation of clinical trials is crucial and should be promoted. Ultimately, a comprehensive strategy integrating pharmacotherapy, personalized medicine, and cutting-edge technologies aims to improve outcomes and enhance the quality of life for individuals with DKD.
Author Contributions
Conceptualization, S.B.N. and S.K.S.; Writing, S.K.S. and S.B.N.; Figures: S.K.S. and S.B.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
All figures were created with BioRender.com, accessed on 12 November 2023.
Conflicts of Interest
S.B.N. reports grant funding from: NIH/NCATS, NIH/NIMHD, CDC, Travere, Bayer; Editorial Board: American Journal of Kidney Disease; Associate Editor, Journal of the American Society of Nephrology; Advisory Board/Steering Committee: Boehringer Ingelheim/Lilly Pharmaceuticals, AstraZeneca, Bayer, NovoNordisk, Janssen Pharmaceutical; Consultant/National Leader: Bayer-ND-CKD; Honoraria: 2021–2022, 2023 Hypertension Highlights, ADA, 2023 ASN nephSAP. S.K.S. has no conflict of interest.
References
- Guariguata, L.; Whiting, D.R.; Hambleton, I.; Beagley, J.; Linnenkamp, U.; Shaw, J.E. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 2014, 103, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; de Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic kidney disease: A report from an ADA Consensus Conference. Diabetes Care 2014, 37, 2864–2883. [Google Scholar] [CrossRef]
- Chen, Y.; Lee, K.; Ni, Z.; He, J.C. Diabetic Kidney Disease: Challenges, Advances, and Opportunities. Kidney Dis. 2020, 6, 215–225. [Google Scholar] [CrossRef]
- Gheith, O.; Farouk, N.; Nampoory, N.; Halim, M.A.; Al-Otaibi, T. Diabetic kidney disease: World wide difference of prevalence and risk factors. J. Nephropharmacol. 2016, 5, 49–56. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Palsson, R.; Patel, U.D. Cardiovascular complications of diabetic kidney disease. Adv. Chronic Kidney Dis. 2014, 21, 273–280. [Google Scholar] [CrossRef]
- Watanabe, K.; Sato, E.; Mishima, E.; Miyazaki, M.; Tanaka, T. What’s New in the Molecular Mechanisms of Diabetic Kidney Disease: Recent Advances. Int. J. Mol. Sci. 2022, 24, 570. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, J.; Wang, W.; An, X.; Luo, L.; Yu, D.; Sun, W. Epigenetic modification in diabetic kidney disease. Front. Endocrinol. 2023, 14, 1133970. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Khan, S.S.; Quaggin, S.E. Therapies on the Horizon for Diabetic Kidney Disease. Curr. Diab. Rep. 2015, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Schnee, J.; Wang, W.; Kim, S.; Fishbein, M.C.; Bruemmer, D.; Law, R.E.; Nicholas, S.; Ross, R.S.; Hsueh, W.A. Osteopontin modulates angiotensin II-induced fibrosis in the intact murine heart. J. Am. Coll. Cardiol. 2004, 43, 1698–1705. [Google Scholar] [CrossRef]
- Wolak, T.; Kim, H.; Ren, Y.; Kim, J.; Vaziri, N.D.; Nicholas, S.B. Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int. 2009, 76, 32–43. [Google Scholar] [CrossRef]
- Nicholas, S.B.; Liu, J.; Kim, J.; Ren, Y.; Collins, A.R.; Nguyen, L.; Hsueh, W.A. Critical role for osteopontin in diabetic nephropathy. Kidney Int. 2010, 77, 588–600. [Google Scholar] [CrossRef]
- Sun, L.; Wu, Y.; Sinha, S.K.; Nicholas, S.B.; Zou, L.X. Performance of multi-biomarker panels based on urinary N-terminal osteopontin for prediction of diabetic kidney disease in patients with diabetes mellitus. Eur. J. Intern. Med. 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Woodhams, L.; Sim, T.F.; Chalmers, L.; Yeap, B.; Green, D.; Schlaich, M.; Schultz, C.; Hillis, G. Diabetic kidney disease in type 2 diabetes: A review of pathogenic mechanisms, patient-related factors and therapeutic options. PeerJ 2021, 9, e11070. [Google Scholar] [CrossRef]
- Matoba, K.; Takeda, Y.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Targeting Redox Imbalance as an Approach for Diabetic Kidney Disease. Biomedicines 2020, 8, 40. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 2000, 28, 1456–1462. [Google Scholar] [CrossRef]
- Ohshiro, Y.; Ma, R.C.; Yasuda, Y.; Hiraoka-Yamamoto, J.; Clermont, A.C.; Isshiki, K.; Yagi, K.; Arikawa, E.; Kern, T.S.; King, G.L. Reduction of diabetes-induced oxidative stress, fibrotic cytokine expression, and renal dysfunction in protein kinase Cbeta-null mice. Diabetes 2006, 55, 3112–3120. [Google Scholar] [CrossRef]
- ElGamal, H.; Munusamy, S. Aldose Reductase as a Drug Target for Treatment of Diabetic Nephropathy: Promises and Challenges. Protein Pept. Lett. 2017, 24, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Menne, J.; Shushakova, N.; Bartels, J.; Kiyan, Y.; Laudeley, R.; Haller, H.; Park, J.K.; Meier, M. Dual inhibition of classical protein kinase C-alpha and protein kinase C-beta isoforms protects against experimental murine diabetic nephropathy. Diabetes 2013, 62, 1167–1174. [Google Scholar] [CrossRef]
- Toyoda, M.; Suzuki, D.; Honma, M.; Uehara, G.; Sakai, T.; Umezono, T.; Sakai, H. High expression of PKC-MAPK pathway mRNAs correlates with glomerular lesions in human diabetic nephropathy. Kidney Int. 2004, 66, 1107–1114. [Google Scholar] [CrossRef]
- Pan, D.; Xu, L.; Guo, M. The role of protein kinase C in diabetic microvascular complications. Front. Endocrinol. 2022, 13, 973058. [Google Scholar] [CrossRef]
- Nobe, K.; Takenouchi, Y.; Kasono, K.; Hashimoto, T.; Honda, K. Two types of overcontraction are involved in intrarenal artery dysfunction in type II diabetic mouse. J. Pharmacol. Exp. Ther. 2014, 351, 77–86. [Google Scholar] [CrossRef]
- Nobe, K.; Nezu, Y.; Tsumita, N.; Hashimoto, T.; Honda, K. Intra- and extrarenal arteries exhibit different profiles of contractile responses in high glucose conditions. Br. J. Pharmacol. 2008, 155, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Ajayi, A.A.; Fidelis, P. The effect of flutamide on systemic and renal hemodynamics in Zucker diabetic rats: Paradoxic renal vasodilator response to endothelin-1 and TXA2 receptor activation in female sex. J. Cardiovasc. Pharmacol. 2006, 48, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Bakris, G.L.; Toto, R.D.; McGill, J.B.; Hu, K.; Anderson, P.W. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care 2005, 28, 2686–2690. [Google Scholar] [CrossRef] [PubMed]
- Thallas-Bonke, V.; Jha, J.C.; Gray, S.P.; Barit, D.; Haller, H.; Schmidt, H.H.; Coughlan, M.T.; Cooper, M.E.; Forbes, J.M.; Jandeleit-Dahm, K.A. Nox-4 deletion reduces oxidative stress and injury by PKC-alpha-associated mechanisms in diabetic nephropathy. Physiol. Rep. 2014, 2, e12192. [Google Scholar] [CrossRef]
- Komeno, M.; Pang, X.; Shimizu, A.; Molla, M.R.; Yasuda-Yamahara, M.; Kume, S.; Rahman, N.I.A.; Soh, J.E.C.; Nguyen, L.K.C.; Ahmat Amin, M.K.B.; et al. Cardio- and reno-protective effects of dipeptidyl peptidase III in diabetic mice. J. Biol. Chem. 2021, 296, 100761. [Google Scholar] [CrossRef]
- Scheinman, J.I.; Fish, A.J.; Matas, A.J.; Michael, A.F. The immunohistopathology of glomerular antigens. II. The glomerular basement membrane, actomyosin, and fibroblast surface antigens in normal, diseased, and transplanted human kidneys. Am. J. Pathol. 1978, 90, 71–88. [Google Scholar]
- Kreisberg, J.I.; Ayo, S.H. The glomerular mesangium in diabetes mellitus. Kidney Int. 1993, 43, 109–113. [Google Scholar] [CrossRef]
- Tahara, A.; Tsukada, J.; Tomura, Y.; Yatsu, T.; Shibasaki, M. Effects of high glucose on AVP-induced hyperplasia, hypertrophy, and type IV collagen synthesis in cultured rat mesangial cells. Endocr. Res. 2012, 37, 216–227. [Google Scholar] [CrossRef]
- Xia, L.; Wang, H.; Goldberg, H.J.; Munk, S.; Fantus, I.G.; Whiteside, C.I. Mesangial cell NADPH oxidase upregulation in high glucose is protein kinase C dependent and required for collagen IV expression. Am. J. Physiol. Renal Physiol. 2006, 290, F345–F356. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Yin, Q. Peroxisome proliferator-activated receptor gamma agonists attenuate hyperglycaemia-induced hyaluronan secretion in vascular smooth muscle cells by inhibiting PKCbeta2. Cell Biochem. Biophys. 2013, 67, 583–590. [Google Scholar] [CrossRef]
- Grigorova-Borsos, A.M.; Bakillah, A.; Urios, P.; Leblond, V.; Guillot, R.; Sternberg, M. Production of type IV collagen and 72-kDa gelatinase by human endothelial cells cultured in high glucose. Effects of a protein kinase C inhibitor, GF 109203X. Biochem. Cell Biol. 1996, 74, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Soetikno, V.; Watanabe, K.; Sari, F.R.; Harima, M.; Thandavarayan, R.A.; Veeraveedu, P.T.; Arozal, W.; Sukumaran, V.; Lakshmanan, A.P.; Arumugam, S.; et al. Curcumin attenuates diabetic nephropathy by inhibiting PKC-alpha and PKC-beta1 activity in streptozotocin-induced type I diabetic rats. Mol. Nutr. Food Res. 2011, 55, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Koya, D.; Jirousek, M.R.; Lin, Y.W.; Ishii, H.; Kuboki, K.; King, G.L. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J. Clin. Investig. 1997, 100, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Mima, A.; Kitada, M.; Geraldes, P.; Li, Q.; Matsumoto, M.; Mizutani, K.; Qi, W.; Li, C.; Leitges, M.; Rask-Madsen, C.; et al. Glomerular VEGF resistance induced by PKCdelta/SHP-1 activation and contribution to diabetic nephropathy. FASEB J. 2012, 26, 2963–2974. [Google Scholar] [CrossRef]
- Meier, M.; Menne, J.; Park, J.K.; Holtz, M.; Gueler, F.; Kirsch, T.; Schiffer, M.; Mengel, M.; Lindschau, C.; Leitges, M.; et al. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J. Am. Soc. Nephrol. 2007, 18, 1190–1198. [Google Scholar] [CrossRef]
- Baccora, M.H.; Cortes, P.; Hassett, C.; Taube, D.W.; Yee, J. Effects of long-term elevated glucose on collagen formation by mesangial cells. Kidney Int. 2007, 72, 1216–1225. [Google Scholar] [CrossRef]
- Whiteside, C.; Wang, H.; Xia, L.; Munk, S.; Goldberg, H.J.; Fantus, I.G. Rosiglitazone prevents high glucose-induced vascular endothelial growth factor and collagen IV expression in cultured mesangial cells. Exp. Diabetes Res. 2009, 2009, 910783. [Google Scholar] [CrossRef]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 187. [Google Scholar] [CrossRef]
- Hathaway, C.K.; Gasim, A.M.; Grant, R.; Chang, A.S.; Kim, H.S.; Madden, V.J.; Bagnell, C.R., Jr.; Jennette, J.C.; Smithies, O.; Kakoki, M. Low TGFbeta1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 5815–5820. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cohen, M.P.; Lautenslager, G.T.; Shearman, C.W.; Ziyadeh, F.N. Glycated albumin stimulates TGF-beta 1 production and protein kinase C activity in glomerular endothelial cells. Kidney Int. 2001, 59, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.F.; Zhou, Q.G.; Hou, F.F.; Liu, B.Y.; Liang, M. Advanced oxidation protein products induce mesangial cell perturbation through PKC-dependent activation of NADPH oxidase. Am. J. Physiol. Renal Physiol. 2009, 296, F427–F437. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Godson, C.; Cannon, S.; Kato, S.; Mackenzie, H.S.; Martin, F.; Brady, H.R. Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J. Biol. Chem. 1999, 274, 5830–5834. [Google Scholar] [CrossRef]
- Xiao, Y.H.; He, X.Y.; Han, Q.; Yang, F.; Zhou, S.X. Atorvastatin prevents glomerular extracellular matrix formation by interfering with the PKC signaling pathway. Mol. Med. Rep. 2018, 17, 6441–6448. [Google Scholar] [CrossRef]
- Liu, H.; Luo, Y.; Zhang, T.; Zhang, Y.; Wu, Q.; Yuan, L.; Chung, S.S.; Oates, P.J.; Yang, J.Y. Genetic deficiency of aldose reductase counteracts the development of diabetic nephropathy in C57BL/6 mice. Diabetologia 2011, 54, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Thallas-Bonke, V.; Banal, C.; Gray, S.P.; Chow, B.S.; Ramm, G.; Quaggin, S.E.; Cooper, M.E.; Schmidt, H.H.; Jandeleit-Dahm, K.A. Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia 2016, 59, 379–389. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.M.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 2019, 62, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Schnaper, H.W. High ambient glucose enhances sensitivity to TGF-beta1 via extracellular signal--regulated kinase and protein kinase Cdelta activities in human mesangial cells. J. Am. Soc. Nephrol. 2004, 15, 2032–2041. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Johnson, E.C.; Cooney, S.K.; Anderberg, R.J.; Johnson, E.K.; Clifton, G.D.; Meek, R.L. Amino acids injure mesangial cells by advanced glycation end products, oxidative stress, and protein kinase C. Kidney Int. 2005, 67, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Peng, F.; Zhang, B.; Ingram, A.J.; Kelly, D.J.; Gilbert, R.E.; Gao, B.; Kumar, S.; Krepinsky, J.C. EGFR-PLCgamma1 signaling mediates high glucose-induced PKCbeta1-Akt activation and collagen I upregulation in mesangial cells. Am. J. Physiol. Renal Physiol. 2009, 297, F822–F834. [Google Scholar] [CrossRef]
- Tokuyama, H.; Kim, S.; Zhang, Y.; Langham, R.G.; Cox, A.J.; Gow, R.M.; Kelly, D.J.; Gilbert, R.E. Protein kinase C beta inhibition ameliorates experimental mesangial proliferative glomerulonephritis. Nephrology 2011, 16, 649–655. [Google Scholar] [CrossRef]
- Garay-Sevilla, M.E.; Rojas, A.; Portero-Otin, M.; Uribarri, J. Dietary AGEs as Exogenous Boosters of Inflammation. Nutrients 2021, 13, 2802. [Google Scholar] [CrossRef]
- Gill, V.; Kumar, V.; Singh, K.; Kumar, A.; Kim, J.J. Advanced Glycation End Products (AGEs) May Be a Striking Link Between Modern Diet and Health. Biomolecules 2019, 9, 888. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, G.E. AGE restriction in diabetes mellitus: A paradigm shift. Nat. Rev. Endocrinol. 2011, 7, 526–539. [Google Scholar] [CrossRef]
- Sergi, D.; Boulestin, H.; Campbell, F.M.; Williams, L.M. The Role of Dietary Advanced Glycation End Products in Metabolic Dysfunction. Mol. Nutr. Food Res. 2021, 65, e1900934. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, E. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J. Inflam. 2013, 2013, 403460. [Google Scholar] [CrossRef]
- Prevost, G.; Fajardy, I.; Besmond, C.; Balkau, B.; Tichet, J.; Fontaine, P.; Danze, P.M.; Marre, M.; Genediab and, D.E.S.I. R Studies. Polymorphisms of the receptor of advanced glycation endproducts (RAGE) and the development of nephropathy in type 1 diabetic patients. Diabetes Metab. 2005, 31, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Keller, J.N. Evaluation of rage isoforms, ligands, and signaling in the brain. Biochim. Biophys. Acta 2005, 1746, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, X.; Tuo, M.; Ma, J.; Xie, A. RAGE and its emerging role in the pathogenesis of Parkinson’s disease. Neurosci. Lett. 2018, 672, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef]
- Sterenczak, K.A.; Nolte, I.; Murua Escobar, H. RAGE splicing variants in mammals. Methods Mol. Biol. 2013, 963, 265–276. [Google Scholar] [CrossRef]
- Bopp, C.; Bierhaus, A.; Hofer, S.; Bouchon, A.; Nawroth, P.P.; Martin, E.; Weigand, M.A. Bench-to-bedside review: The inflammation-perpetuating pattern-recognition receptor RAGE as a therapeutic target in sepsis. Crit. Care 2008, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.; Yan, S.D.; Yan, S.F.; Schmidt, A.M. Receptor for advanced glycation endproducts: A multiligand receptor magnifying cell stress in diverse pathologic settings. Adv. Drug Deliv. Rev. 2002, 54, 1615–1625. [Google Scholar] [CrossRef]
- Gasiorowski, K.; Brokos, B.; Echeverria, V.; Barreto, G.E.; Leszek, J. RAGE-TLR Crosstalk Sustains Chronic Inflammation in Neurodegeneration. Mol. Neurobiol. 2018, 55, 1463–1476. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Naka, Y.; Schmidt, A.M. Glycation, inflammation, and RAGE: A scaffold for the macrovascular complications of diabetes and beyond. Circ. Res. 2003, 93, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Kass, D.A.; Shapiro, E.P.; Kawaguchi, M.; Capriotti, A.R.; Scuteri, A.; deGroof, R.C.; Lakatta, E.G. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation 2001, 104, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.I.; Wuerth, J.P.; Cartwright, K.; Bain, R.P.; Dippe, S.; Hershon, K.; Mooradian, A.D.; Spinowitz, B.S. Design and baseline characteristics for the aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II). Control Clin. Trials 1999, 20, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, E.D.; Weigert, C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int. Suppl. 2000, 77, S13–S18. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [CrossRef] [PubMed]
- Kolm-Litty, V.; Sauer, U.; Nerlich, A.; Lehmann, R.; Schleicher, E.D. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. J. Clin. Investig. 1998, 101, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Beriault, D.R.; Werstuck, G.H. The role of glucosamine-induced ER stress in diabetic atherogenesis. Exp. Diabetes Res. 2012, 2012, 187018. [Google Scholar] [CrossRef]
- Marsh, S.A.; Dell’Italia, L.J.; Chatham, J.C. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids 2011, 40, 819–828. [Google Scholar] [CrossRef]
- Taparra, K.; Tran, P.T.; Zachara, N.E. Hijacking the Hexosamine Biosynthetic Pathway to Promote EMT-Mediated Neoplastic Phenotypes. Front. Oncol. 2016, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, S.B.; Aguiniga, E.; Ren, Y.; Kim, J.; Wong, J.; Govindarajan, N.; Noda, M.; Wang, W.; Kawano, Y.; Collins, A.; et al. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int. 2005, 67, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Chiaradonna, F.; Ricciardiello, F.; Palorini, R. The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 2018, 7, 53. [Google Scholar] [CrossRef]
- Pang, Y.; Bounelis, P.; Chatham, J.C.; Marchase, R.B. Hexosamine pathway is responsible for inhibition by diabetes of phenylephrine-induced inotropy. Diabetes 2004, 53, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 2003, 14, S233–S236. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Model. Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef]
- Yabe-Nishimura, C. Aldose reductase in glucose toxicity: A potential target for the prevention of diabetic complications. Pharmacol. Rev. 1998, 50, 21–33. [Google Scholar]
- Yan, L.J. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. J. Diabetes Res. 2014, 2014, 137919. [Google Scholar] [CrossRef]
- Dunlop, M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int. Suppl. 2000, 77, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Demir, Y.; Isik, M.; Gulcin, I.; Beydemir, S. Phenolic compounds inhibit the aldose reductase enzyme from the sheep kidney. J. Biochem. Mol. Toxicol. 2017, 31, e21936. [Google Scholar] [CrossRef]
- Chitra, P.S.; Chaki, D.; Boiroju, N.K.; Mokalla, T.R.; Gadde, A.K.; Agraharam, S.G.; Reddy, G.B. Status of oxidative stress markers, advanced glycation index, and polyol pathway in age-related cataract subjects with and without diabetes. Exp. Eye Res. 2020, 200, 108230. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and localization of the principal fructose-metabolizing enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef]
- Baeza, J.; Smallegan, M.J.; Denu, J.M. Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 2015, 10, 122–128. [Google Scholar] [CrossRef]
- Forbes, J.M. Mitochondria-Power Players in Kidney Function? Trends Endocrinol. Metab. 2016, 27, 441–442. [Google Scholar] [CrossRef]
- Xu, J.; Kitada, M.; Koya, D. NAD(+) Homeostasis in Diabetic Kidney Disease. Front. Med. 2021, 8, 703076. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R. Redox imbalance in diabetes. Antioxid. Redox Signal. 2007, 9, 865–867. [Google Scholar] [CrossRef]
- Wu, J.; Jin, Z.; Zheng, H.; Yan, L.J. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab. Syndr. Obes. 2016, 9, 145–153. [Google Scholar] [CrossRef]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Diaz, A.G.; Pazarin-Villasenor, L.; Yanowsky-Escatell, F.G.; Andrade-Sierra, J. Oxidative Stress in Diabetic Nephropathy with Early Chronic Kidney Disease. J. Diabetes Res. 2016, 2016, 7047238. [Google Scholar] [CrossRef] [PubMed]
- Tilton, R.G.; Baier, L.D.; Harlow, J.E.; Smith, S.R.; Ostrow, E.; Williamson, J.R. Diabetes-induced glomerular dysfunction: Links to a more reduced cytosolic ratio of NADH/NAD+. Kidney Int. 1992, 41, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, R.; Yan, L.J. Roles of Pyruvate, NADH, and Mitochondrial Complex I in Redox Balance and Imbalance in beta Cell Function and Dysfunction. J. Diabetes Res. 2015, 2015, 512618. [Google Scholar] [CrossRef]
- Zakaria, E.M.; El-Maraghy, N.N.; Ahmed, A.F.; Ali, A.A.; El-Bassossy, H.M. PARP inhibition ameliorates nephropathy in an animal model of type 2 diabetes: Focus on oxidative stress, inflammation, and fibrosis. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jin, Z.; Yan, L.J. Redox imbalance and mitochondrial abnormalities in the diabetic lung. Redox Biol. 2017, 11, 51–59. [Google Scholar] [CrossRef]
- Benzi, A.; Sturla, L.; Heine, M.; Fischer, A.W.; Spinelli, S.; Magnone, M.; Sociali, G.; Parodi, A.; Fenoglio, D.; Emionite, L.; et al. CD38 downregulation modulates NAD(+) and NADP(H) levels in thermogenic adipose tissues. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158819. [Google Scholar] [CrossRef]
- Masutani, M.; Suzuki, H.; Kamada, N.; Watanabe, M.; Ueda, O.; Nozaki, T.; Jishage, K.; Watanabe, T.; Sugimoto, T.; Nakagama, H.; et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 2301–2304. [Google Scholar] [CrossRef]
- Shi, B.; Wang, W.; Korman, B.; Kai, L.; Wang, Q.; Wei, J.; Bale, S.; Marangoni, R.G.; Bhattacharyya, S.; Miller, S.; et al. Targeting CD38-dependent NAD(+) metabolism to mitigate multiple organ fibrosis. iScience 2021, 24, 101902. [Google Scholar] [CrossRef] [PubMed]
- Van Buren, P.N.; Toto, R. Hypertension in diabetic nephropathy: Epidemiology, mechanisms, and management. Adv. Chronic. Kidney Dis. 2011, 18, 28–41. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Magee, G.M.; Bilous, R.W.; Cardwell, C.R.; Hunter, S.J.; Kee, F.; Fogarty, D.G. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 2009, 52, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Jerums, G.; Premaratne, E.; Panagiotopoulos, S.; MacIsaac, R.J. The clinical significance of hyperfiltration in diabetes. Diabetologia 2010, 53, 2093–2104. [Google Scholar] [CrossRef]
- Tonneijck, L.; Muskiet, M.H.; Smits, M.M.; van Bommel, E.J.; Heerspink, H.J.; van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Lawler, E.V.; Mackenzie, H.S. The hyperfiltration theory: A paradigm shift in nephrology. Kidney Int. 1996, 49, 1774–1777. [Google Scholar] [CrossRef]
- Vallon, V.; Komers, R. Pathophysiology of the diabetic kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar] [CrossRef]
- Vallon, V.; Huang, D.Y.; Deng, A.; Richter, K.; Blantz, R.C.; Thomson, S. Salt-sensitivity of proximal reabsorption alters macula densa salt and explains the paradoxical effect of dietary salt on glomerular filtration rate in diabetes mellitus. J. Am. Soc. Nephrol. 2002, 13, 1865–1871. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Furuki, T.; Kobori, H.; Miyakawa, M.; Imachi, H.; Murao, K.; Nishiyama, A. Effects of sodium-glucose cotransporter 2 inhibitors on urinary excretion of intact and total angiotensinogen in patients with type 2 diabetes. J. Investig. Med. 2017, 65, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.J.; Chung, S.; Kim, S.J.; Lee, E.M.; Yoo, Y.H.; Kim, J.W.; Ahn, Y.B.; Kim, E.S.; Moon, S.D.; Kim, M.J.; et al. Effect of Sodium-Glucose Co-Transporter 2 Inhibitor, Dapagliflozin, on Renal Renin-Angiotensin System in an Animal Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0165703. [Google Scholar] [CrossRef]
- Velez, J.C. The importance of the intrarenal renin-angiotensin system. Nat. Clin. Pract. Nephrol. 2009, 5, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Roscioni, S.S.; Heerspink, H.J.; de Zeeuw, D. The effect of RAAS blockade on the progression of diabetic nephropathy. Nat. Rev. Nephrol. 2014, 10, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S.; et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Price, D.A.; Porter, L.E.; Gordon, M.; Fisher, N.D.; De’Oliveira, J.M.; Laffel, L.M.; Passan, D.R.; Williams, G.H.; Hollenberg, N.K. The paradox of the low-renin state in diabetic nephropathy. J. Am. Soc. Nephrol. 1999, 10, 2382–2391. [Google Scholar] [CrossRef]
- Gurley, S.B.; Coffman, T.M. The renin-angiotensin system and diabetic nephropathy. Semin. Nephrol. 2007, 27, 144–152. [Google Scholar] [CrossRef]
- Lai, K.N.; Leung, J.C.; Lai, K.B.; To, W.Y.; Yeung, V.T.; Lai, F.M. Gene expression of the renin-angiotensin system in human kidney. J. Hypertens. 1998, 16, 91–102. [Google Scholar] [CrossRef]
- Kaneshiro, Y.; Ichihara, A.; Sakoda, M.; Takemitsu, T.; Nabi, A.H.; Uddin, M.N.; Nakagawa, T.; Nishiyama, A.; Suzuki, F.; Inagami, T.; et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J. Am. Soc. Nephrol. 2007, 18, 1789–1795. [Google Scholar] [CrossRef]
- Peters, B.; Grisk, O.; Becher, B.; Wanka, H.; Kuttler, B.; Ludemann, J.; Lorenz, G.; Rettig, R.; Mullins, J.J.; Peters, J. Dose-dependent titration of prorenin and blood pressure in Cyp1a1ren-2 transgenic rats: Absence of prorenin-induced glomerulosclerosis. J. Hypertens. 2008, 26, 102–109. [Google Scholar] [CrossRef]
- Zimpelmann, J.; Kumar, D.; Levine, D.Z.; Wehbi, G.; Imig, J.D.; Navar, L.G.; Burns, K.D. Early diabetes mellitus stimulates proximal tubule renin mRNA expression in the rat. Kidney Int. 2000, 58, 2320–2330. [Google Scholar] [CrossRef]
- Hsieh, T.J.; Zhang, S.L.; Filep, J.G.; Tang, S.S.; Ingelfinger, J.R.; Chan, J.S. High glucose stimulates angiotensinogen gene expression via reactive oxygen species generation in rat kidney proximal tubular cells. Endocrinology 2002, 143, 2975–2985. [Google Scholar] [CrossRef]
- Nguyen, G.; Muller, D.N. The biology of the (pro)renin receptor. J. Am. Soc. Nephrol. 2010, 21, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.; Yusuf, S.; Mann, J.F.E.; Hoogwerf, B.; Zinman, B.; Held, C.; Fisher, M.; Wolffenbuttel, B.H.R.; Pagans, J.B.; Richardson, L.; et al. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: Results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000, 355, 253–259. [Google Scholar]
- Casas, J.P.; Chua, W.; Loukogeorgakis, S.; Vallance, P.; Smeeth, L.; Hingorani, A.D.; MacAllister, R.J. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: Systematic review and meta-analysis. Lancet 2005, 366, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Barnett, A.H.; Bain, S.C.; Bouter, P.; Karlberg, B.; Madsbad, S.; Jervell, J.; Mustonen, J.; Diabetics Exposed to Telmisartan and Enalapril Study Group. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N. Engl. J. Med. 2004, 351, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Jennings, D.L.; Kalus, J.S.; Coleman, C.I.; Manierski, C.; Yee, J. Combination therapy with an ACE inhibitor and an angiotensin receptor blocker for diabetic nephropathy: A meta-analysis. Diabet. Med. 2007, 24, 486–493. [Google Scholar] [CrossRef]
- Banerjee, D.; Winocour, P.; Chowdhury, T.A.; De, P.; Wahba, M.; Montero, R.; Fogarty, D.; Frankel, A.H.; Karalliedde, J.; Mark, P.B.; et al. Management of hypertension and renin-angiotensin-aldosterone system blockade in adults with diabetic kidney disease: Association of British Clinical Diabetologists and the Renal Association UK guideline update 2021. BMC Nephrol. 2022, 23, 9. [Google Scholar] [CrossRef]
- Leehey, D.J.; Zhang, J.H.; Emanuele, N.V.; Whaley-Connell, A.; Palevsky, P.M.; Reilly, R.F.; Guarino, P.; Fried, L.F.; Group, V.N.-D.S. BP and Renal Outcomes in Diabetic Kidney Disease: The Veterans Affairs Nephropathy in Diabetes Trial. Clin. J. Am. Soc. Nephrol. 2015, 10, 2159–2169. [Google Scholar] [CrossRef]
- Hollenberg, N.K. Direct renin inhibition and the kidney. Nat. Rev. Nephrol. 2010, 6, 49–55. [Google Scholar] [CrossRef]
- Huang, Y.; Wongamorntham, S.; Kasting, J.; McQuillan, D.; Owens, R.T.; Yu, L.; Noble, N.A.; Border, W. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. 2006, 69, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Noble, N.A.; Zhang, J.; Xu, C.; Border, W.A. Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int. 2007, 72, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sakoda, M.; Ichihara, A.; Kaneshiro, Y.; Takemitsu, T.; Nakazato, Y.; Nabi, A.H.; Nakagawa, T.; Suzuki, F.; Inagami, T.; Itoh, H. (Pro)renin receptor-mediated activation of mitogen-activated protein kinases in human vascular smooth muscle cells. Hypertens. Res. 2007, 30, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Kaneshiro, Y.; Ichihara, A.; Takemitsu, T.; Sakoda, M.; Suzuki, F.; Nakagawa, T.; Hayashi, M.; Inagami, T. Increased expression of cyclooxygenase-2 in the renal cortex of human prorenin receptor gene-transgenic rats. Kidney Int. 2006, 70, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Feldt, S.; Batenburg, W.W.; Mazak, I.; Maschke, U.; Wellner, M.; Kvakan, H.; Dechend, R.; Fiebeler, A.; Burckle, C.; Contrepas, A.; et al. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension 2008, 51, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Mercure, C.; Prescott, G.; Lacombe, M.J.; Silversides, D.W.; Reudelhuber, T.L. Chronic increases in circulating prorenin are not associated with renal or cardiac pathologies. Hypertension 2009, 53, 1062–1069. [Google Scholar] [CrossRef]
- Danser, A.H. (Pro)renin receptor and vacuolar H+-ATPase. Hypertension 2009, 54, 219–221. [Google Scholar] [CrossRef]
- Advani, A.; Kelly, D.J.; Cox, A.J.; White, K.E.; Advani, S.L.; Thai, K.; Connelly, K.A.; Yuen, D.; Trogadis, J.; Herzenberg, A.M.; et al. The (Pro)renin receptor: Site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension 2009, 54, 261–269. [Google Scholar] [CrossRef]
- Luetscher, J.A.; Kraemer, F.B.; Wilson, D.M.; Schwartz, H.C.; Bryer-Ash, M. Increased plasma inactive renin in diabetes mellitus. A marker of microvascular complications. N. Engl. J. Med. 1985, 312, 1412–1417. [Google Scholar] [CrossRef]
- Siragy, H.M.; Huang, J. Renal (pro)renin receptor upregulation in diabetic rats through enhanced angiotensin AT1 receptor and NADPH oxidase activity. Exp. Physiol. 2008, 93, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Border, W.A.; Noble, N.A. Functional renin receptors in renal mesangial cells. Curr. Hypertens. Rep. 2007, 9, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.; Delarue, F.; Burckle, C.; Bouzhir, L.; Giller, T.; Sraer, J.D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Investig. 2002, 109, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Durvasula, R.V.; Shankland, S.J. Activation of a local renin angiotensin system in podocytes by glucose. Am. J. Physiol. Renal Physiol. 2008, 294, F830–F839. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Jung, F.F.; Ingelfinger, J.R. Renal renin-angiotensin system in diabetes: Functional, immunohistochemical, and molecular biological correlations. Am. J. Physiol. 1993, 265, F477–F486. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.Y.; Cogan, M.G. Angiotensin II stimulates early proximal bicarbonate absorption in the rat by decreasing cyclic adenosine monophosphate. J. Clin. Investig. 1989, 84, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.Y.; Cogan, M.G. Role of protein kinase C in proximal bicarbonate absorption and angiotensin signaling. Am. J. Physiol. 1990, 258, F927–F933. [Google Scholar] [CrossRef]
- Kifor, I.; Moore, T.J.; Fallo, F.; Sperling, E.; Chiou, C.Y.; Menachery, A.; Williams, G.H. Potassium-stimulated angiotensin release from superfused adrenal capsules and enzymatically dispersed cells of the zona glomerulosa. Endocrinology 1991, 129, 823–831. [Google Scholar] [CrossRef]
- Singh, R.; Alavi, N.; Singh, A.K.; Leehey, D.J. Role of angiotensin II in glucose-induced inhibition of mesangial matrix degradation. Diabetes 1999, 48, 2066–2073. [Google Scholar] [CrossRef]
- Mezzano, S.A.; Ruiz-Ortega, M.; Egido, J. Angiotensin II and renal fibrosis. Hypertension 2001, 38, 635–638. [Google Scholar] [CrossRef]
- Premilovac, D.; Attrill, E.; Rattigan, S.; Richards, S.M.; Kim, J.; Keske, M.A. Acute, local infusion of angiotensin II impairs microvascular and metabolic insulin sensitivity in skeletal muscle. Cardiovasc. Res. 2019, 115, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Velloso, L.A.; Folli, F.; Perego, L.; Saad, M.J. The multi-faceted cross-talk between the insulin and angiotensin II signaling systems. Diabetes Metab. Res. Rev. 2006, 22, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Halagappa, V.K.; Riazi, S.; Hu, X.; Ecelbarger, C.A. Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J. Am. Soc. Nephrol. 2007, 18, 2661–2671. [Google Scholar] [CrossRef]
- Mezzano, S.; Droguett, A.; Burgos, M.E.; Ardiles, L.G.; Flores, C.A.; Aros, C.A.; Caorsi, I.; Vio, C.P.; Ruiz-Ortega, M.; Egido, J. Renin-angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int. Suppl. 2003, 86, S64–S70. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhou, L. The Signaling of Cellular Senescence in Diabetic Nephropathy. Oxid. Med. Cell Longev. 2019, 2019, 7495629. [Google Scholar] [CrossRef]
- Araujo, L.S.; Torquato, B.G.S.; da Silva, C.A.; Dos Reis Monteiro, M.L.G.; Dos Santos Martins, A.L.M.; da Silva, M.V.; Dos Reis, M.A.; Machado, J.R. Renal expression of cytokines and chemokines in diabetic nephropathy. BMC Nephrol. 2020, 21, 308. [Google Scholar] [CrossRef]
- Salti, T.; Khazim, K.; Haddad, R.; Campisi-Pinto, S.; Bar-Sela, G.; Cohen, I. Glucose Induces IL-1alpha-Dependent Inflammation and Extracellular Matrix Proteins Expression and Deposition in Renal Tubular Epithelial Cells in Diabetic Kidney Disease. Front. Immunol. 2020, 11, 1270. [Google Scholar] [CrossRef] [PubMed]
- Milas, O.; Gadalean, F.; Vlad, A.; Dumitrascu, V.; Velciov, S.; Gluhovschi, C.; Bob, F.; Popescu, R.; Ursoniu, S.; Jianu, D.C.; et al. Pro-inflammatory cytokines are associated with podocyte damage and proximal tubular dysfunction in the early stage of diabetic kidney disease in type 2 diabetes mellitus patients. J. Diabetes Complicat. 2020, 34, 107479. [Google Scholar] [CrossRef]
- Sangoi, M.B.; de Carvalho, J.A.; Tatsch, E.; Hausen, B.S.; Bollick, Y.S.; Londero, S.W.; Duarte, T.; Scolari, R.; Duarte, M.M.; Premaor, M.O.; et al. Urinary inflammatory cytokines as indicators of kidney damage in type 2 diabetic patients. Clin. Chim. Acta 2016, 460, 178–183. [Google Scholar] [CrossRef]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Dalla Vestra, M.; Mussap, M.; Gallina, P.; Bruseghin, M.; Cernigoi, A.M.; Saller, A.; Plebani, M.; Fioretto, P. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J. Am. Soc. Nephrol. 2005, 16 (Suppl. S1), S78–S82. [Google Scholar] [CrossRef]
- Suzuki, D.; Miyazaki, M.; Naka, R.; Koji, T.; Yagame, M.; Jinde, K.; Endoh, M.; Nomoto, Y.; Sakai, H. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes 1995, 44, 1233–1238. [Google Scholar] [CrossRef]
- Shikano, M.; Sobajima, H.; Yoshikawa, H.; Toba, T.; Kushimoto, H.; Katsumata, H.; Tomita, M.; Kawashima, S. Usefulness of a highly sensitive urinary and serum IL-6 assay in patients with diabetic nephropathy. Nephron 2000, 85, 81–85. [Google Scholar] [CrossRef]
- McGeough, M.D.; Wree, A.; Inzaugarat, M.E.; Haimovich, A.; Johnson, C.D.; Pena, C.A.; Goldbach-Mansky, R.; Broderick, L.; Feldstein, A.E.; Hoffman, H.M. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J. Clin. Investig. 2017, 127, 4488–4497. [Google Scholar] [CrossRef]
- Amaral, F.A.; Bastos, L.F.; Oliveira, T.H.; Dias, A.C.; Oliveira, V.L.; Tavares, L.D.; Costa, V.V.; Galvao, I.; Soriani, F.M.; Szymkowski, D.E.; et al. Transmembrane TNF-alpha is sufficient for articular inflammation and hypernociception in a mouse model of gout. Eur. J. Immunol. 2016, 46, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Nadkarni, G.N.; Huang, Y.; Moledina, D.G.; Rao, V.; Zhang, J.; Ferket, B.; Crowley, S.T.; Fried, L.F.; Parikh, C.R. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2786–2793. [Google Scholar] [CrossRef] [PubMed]
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Brian Reeves, W. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Liang, R.; Huang, B.; Hou, J.; Yin, J.; Zhao, T.; Zhou, L.; Wu, R.; Qian, Y.; Wang, F. Tumor necrosis factor-alpha blockade ameliorates diabetic nephropathy in rats. Clin. Kidney J. 2021, 14, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.J.; Yang, H.Y.; Pai, M.H.; Wu, C.H.; Chen, J.R. Long-term administration of advanced glycation end-product stimulates the activation of NLRP3 inflammasome and sparking the development of renal injury. J. Nutr. Biochem. 2017, 39, 68–76. [Google Scholar] [CrossRef]
- Bonner, R.; Albajrami, O.; Hudspeth, J.; Upadhyay, A. Diabetic Kidney Disease. Prim. Care 2020, 47, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.; Zoungas, S.; Rossing, P.; Groop, P.H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef]
- Flyvbjerg, A. The role of the complement system in diabetic nephropathy. Nat. Rev. Nephrol. 2017, 13, 311–318. [Google Scholar] [CrossRef]
- Qian, Y.; Feldman, E.; Pennathur, S.; Kretzler, M.; Brosius, F.C., 3rd. From fibrosis to sclerosis: Mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes 2008, 57, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Najafian, B.; Alpers, C.E.; Fogo, A.B. Pathology of human diabetic nephropathy. Contrib. Nephrol. 2011, 170, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Bulow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Menn-Josephy, H.; Lee, C.S.; Nolin, A.; Christov, M.; Rybin, D.V.; Weinberg, J.M.; Henderson, J.; Bonegio, R.; Havasi, A. Renal Interstitial Fibrosis: An Imperfect Predictor of Kidney Disease Progression in Some Patient Cohorts. Am. J. Nephrol. 2016, 44, 289–299. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Essawy, M.; Soylemezoglu, O.; Muchaneta-Kubara, E.C.; Shortland, J.; Brown, C.B.; el Nahas, A.M. Myofibroblasts and the progression of diabetic nephropathy. Nephrol. Dial. Transplant. 1997, 12, 43–50. [Google Scholar] [CrossRef]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, D.; Kang, X.; Zhou, R.; Sun, Y.; Lian, F.; Tong, X. Signaling Pathways Involved in Diabetic Renal Fibrosis. Front. Cell Dev. Biol. 2021, 9, 696542. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.L.; Liu, T.T.; Lan, H.Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 7881. [Google Scholar] [CrossRef]
- Kayhan, M.; Vouillamoz, J.; Rodriguez, D.G.; Bugarski, M.; Mitamura, Y.; Gschwend, J.; Schneider, C.; Hall, A.; Legouis, D.; Akdis, C.A.; et al. Intrinsic TGF-beta signaling attenuates proximal tubule mitochondrial injury and inflammation in chronic kidney disease. Nat. Commun. 2023, 14, 3236. [Google Scholar] [CrossRef]
- Hoffman, B.B.; Sharma, K.; Zhu, Y.; Ziyadeh, F.N. Transcriptional activation of transforming growth factor-beta1 in mesangial cell culture by high glucose concentration. Kidney Int. 1998, 54, 1107–1116. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-beta activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Skorczewski, J.; Feng, X.; Mei, L.; Murphy-Ullrich, J.E. Glucose up-regulates thrombospondin 1 gene transcription and transforming growth factor-beta activity through antagonism of cGMP-dependent protein kinase repression via upstream stimulatory factor 2. J. Biol. Chem. 2004, 279, 34311–34322. [Google Scholar] [CrossRef]
- Isono, M.; Mogyorosi, A.; Han, D.C.; Hoffman, B.B.; Ziyadeh, F.N. Stimulation of TGF-beta type II receptor by high glucose in mouse mesangial cells and in diabetic kidney. Am. J. Physiol. Renal Physiol. 2000, 278, F830–F838. [Google Scholar] [CrossRef]
- Isono, M.; Chen, S.; Hong, S.W.; Iglesias-de la Cruz, M.C.; Ziyadeh, F.N. Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-beta-induced fibronectin in mesangial cells. Biochem. Biophys. Res. Commun. 2002, 296, 1356–1365. [Google Scholar] [CrossRef]
- Hong, S.W.; Isono, M.; Chen, S.; Iglesias-De La Cruz, M.C.; Han, D.C.; Ziyadeh, F.N. Increased glomerular and tubular expression of transforming growth factor-beta1, its type II receptor, and activation of the Smad signaling pathway in the db/db mouse. Am. J. Pathol. 2001, 158, 1653–1663. [Google Scholar] [CrossRef]
- Lan, H.Y.; Chung, A.C.K. Transforming growth factor-beta and Smads. Contrib. Nephrol. 2011, 170, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-De La Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef] [PubMed]
- Baricos, W.H.; Cortez, S.L.; Deboisblanc, M.; Xin, S. Transforming growth factor-beta is a potent inhibitor of extracellular matrix degradation by cultured human mesangial cells. J. Am. Soc. Nephrol. 1999, 10, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Solbes, A.S.; Youker, K. Epithelial to Mesenchymal Transition (EMT) and Endothelial to Mesenchymal Transition (EndMT): Role and Implications in Kidney Fibrosis. Results Probl. Cell Differ. 2017, 60, 345–372. [Google Scholar] [CrossRef]
- Sharma, K.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; et al. Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1144–1151. [Google Scholar] [CrossRef]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-beta1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef]
- Haneda, M.; Araki, S.; Togawa, M.; Sugimoto, T.; Isono, M.; Kikkawa, R. Mitogen-activated protein kinase cascade is activated in glomeruli of diabetic rats and glomerular mesangial cells cultured under high glucose conditions. Diabetes 1997, 46, 847–853. [Google Scholar] [CrossRef]
- Li, Z.; Liu, X.; Wang, B.; Nie, Y.; Wen, J.; Wang, Q.; Gu, C. Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology 2017, 22, 589–597. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, H.B.; Lin, Y.; Yao, W.; Huang, J.Y. KLF15 suppresses cell proliferation and extracellular matrix expression in mesangial cells under high glucose. Int. J. Clin. Exp. Med. 2015, 8, 20330–20336. [Google Scholar]
- Wada, T.; Azuma, H.; Furuichi, K.; Sakai, N.; Kitagawa, K.; Iwata, Y.; Matsushima, K.; Takahara, S.; Yokoyama, H.; Kaneko, S. Reduction in chronic allograft nephropathy by inhibition of p38 mitogen-activated protein kinase. Am. J. Nephrol. 2006, 26, 319–325. [Google Scholar] [CrossRef]
- Nishida, M.; Okumura, Y.; Sato, H.; Hamaoka, K. Delayed inhibition of p38 mitogen-activated protein kinase ameliorates renal fibrosis in obstructive nephropathy. Nephrol. Dial. Transplant. 2008, 23, 2520–2524. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.Y.; Flanc, R.S.; Tesch, G.H.; Han, Y.; Atkins, R.C.; Bennett, B.L.; Friedman, G.C.; Fan, J.H.; Nikolic-Paterson, D.J. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J. Am. Soc. Nephrol. 2007, 18, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Yuan, Y.; Zhang, T.; Xu, B.; Gao, Q.; Guan, T. Pentosan polysulfate ameliorates apoptosis and inflammation by suppressing activation of the p38 MAPK pathway in high glucose-treated HK-2 cells. Int. J. Mol. Med. 2018, 41, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, R.; Wu, X.; Chen, Y.; Ji, W.; Wang, J.; Zhang, Y.; Xia, Y.; Tang, Y.; Yuan, J. The Wnt Signaling Pathway in Diabetic Nephropathy. Front. Cell Dev. Biol. 2021, 9, 701547. [Google Scholar] [CrossRef]
- Nie, X.; Wei, X.; Ma, H.; Fan, L.; Chen, W.D. The complex role of Wnt ligands in type 2 diabetes mellitus and related complications. J. Cell Mol. Med. 2021, 25, 6479–6495. [Google Scholar] [CrossRef]
- Zuo, Y.; Liu, Y. New insights into the role and mechanism of Wnt/beta-catenin signalling in kidney fibrosis. Nephrology 2018, 23 (Suppl. S4), 38–43. [Google Scholar] [CrossRef]
- Malik, S.A.; Modarage, K.; Goggolidou, P. The Role of Wnt Signalling in Chronic Kidney Disease (CKD). Genes 2020, 11, 496. [Google Scholar] [CrossRef]
- Guo, Q.; Zhong, W.; Duan, A.; Sun, G.; Cui, W.; Zhuang, X.; Liu, L. Protective or deleterious role of Wnt/beta-catenin signaling in diabetic nephropathy: An unresolved issue. Pharmacol. Res. 2019, 144, 151–157. [Google Scholar] [CrossRef]
- Babayeva, S.; Zilber, Y.; Torban, E. Planar cell polarity pathway regulates actin rearrangement, cell shape, motility, and nephrin distribution in podocytes. Am. J. Physiol. Renal Physiol. 2011, 300, F549–F560. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Stolz, D.B.; Kiss, L.P.; Monga, S.P.; Holzman, L.B.; Liu, Y. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J. Am. Soc. Nephrol. 2009, 20, 1997–2008. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; He, W.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Liu, Y. Mutual antagonism of Wilms’ tumor 1 and beta-catenin dictates podocyte health and disease. J. Am. Soc. Nephrol. 2015, 26, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wan, J.; Hou, X.; Geng, J.; Li, X.; Bai, X. MicroRNA-27a promotes podocyte injury via PPARgamma-mediated beta-catenin activation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2658. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Wang, J.Y.; Ko, J.Y.; Huang, Y.T.; Kuo, Y.H.; Wang, F.S. Dickkopf-1 promotes hyperglycemia-induced accumulation of mesangial matrix and renal dysfunction. J. Am. Soc. Nephrol. 2010, 21, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Wang, M.; Yang, S.; Liu, F.; Sun, L. A glimpse of the pathogenetic mechanisms of Wnt/beta-catenin signaling in diabetic nephropathy. Biomed Res. Int. 2013, 2013, 987064. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Wang, J.Y.; Huang, Y.T.; Kuo, Y.H.; Surendran, K.; Wang, F.S. Wnt/beta-catenin signaling modulates survival of high glucose-stressed mesangial cells. J. Am. Soc. Nephrol. 2006, 17, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.W.; Hsu, Y.C.; Shih, Y.H.; Chang, P.J.; Lin, C.L. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology 2018, 23 (Suppl. S4), 32–37. [Google Scholar] [CrossRef]
- Lin, C.L.; Cheng, H.; Tung, C.W.; Huang, W.J.; Chang, P.J.; Yang, J.T.; Wang, J.Y. Simvastatin reverses high glucose-induced apoptosis of mesangial cells via modulation of Wnt signaling pathway. Am. J. Nephrol. 2008, 28, 290–297. [Google Scholar] [CrossRef]
- Zhu, D.; Yu, H.; He, H.; Ding, J.; Tang, J.; Cao, D.; Hao, L. Spironolactone inhibits apoptosis in rat mesangial cells under hyperglycaemic conditions via the Wnt signalling pathway. Mol. Cell Biochem. 2013, 380, 185–193. [Google Scholar] [CrossRef]
- Hao, J.; Li, F.; Liu, W.; Liu, Q.; Liu, S.; Li, H.; Duan, H. Phosphorylation of PRAS40-Thr246 involved in renal lipid accumulation of diabetes. J. Cell Physiol. 2014, 229, 1069–1077. [Google Scholar] [CrossRef]
- Xue, M.; Cheng, Y.; Han, F.; Chang, Y.; Yang, Y.; Li, X.; Chen, L.; Lu, Y.; Sun, B.; Chen, L. Triptolide Attenuates Renal Tubular Epithelial-mesenchymal Transition Via the MiR-188-5p-mediated PI3K/AKT Pathway in Diabetic Kidney Disease. Int. J. Biol. Sci. 2018, 14, 1545–1557. [Google Scholar] [CrossRef]
- Xu, Z.; Jia, K.; Wang, H.; Gao, F.; Zhao, S.; Li, F.; Hao, J. METTL14-regulated PI3K/Akt signaling pathway via PTEN affects HDAC5-mediated epithelial-mesenchymal transition of renal tubular cells in diabetic kidney disease. Cell Death Dis. 2021, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wen, X.; Zhang, Z.; Xie, M.; Zhou, J. Phillyrin ameliorates diabetic nephropathy through the PI3K/Akt/GSK-3beta signalling pathway in streptozotocin-induced diabetic mice. Hum. Exp. Toxicol. 2021, 40, S487–S496. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jin, M.; Zhao, X.; Zhao, T.; Lin, W.; He, Z.; Fan, M.; Jin, W.; Zhou, J.; Jin, L.; et al. FGF1(DeltaHBS) ameliorates chronic kidney disease via PI3K/AKT mediated suppression of oxidative stress and inflammation. Cell Death Dis. 2019, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.H.; Huang, Y.; Li, W.Q.; Chen, R.G. Influence of LncRNA UCA1 on glucose metabolism in rats with diabetic nephropathy through PI3K-Akt signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10058–10064. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, J.; Cao, Y.; Zhang, R.; Wang, Y.; Zhang, S.; Dai, W.; Ye, S. Silibinin ameliorates diabetic nephropathy via improving diabetic condition in the mice. Eur. J. Pharmacol. 2019, 845, 24–31. [Google Scholar] [CrossRef]
- Jing, D.; Bai, H.; Yin, S. Renoprotective effects of emodin against diabetic nephropathy in rat models are mediated via PI3K/Akt/GSK-3beta and Bax/caspase-3 signaling pathways. Exp. Ther. Med. 2017, 14, 5163–5169. [Google Scholar] [CrossRef]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3beta in Regulating Mitochondrial Activity. Cell Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Inhibitory Effect of Astaxanthin on Oxidative Stress-Induced Mitochondrial Dysfunction—A Mini-Review. Nutrients 2018, 10, 1137. [Google Scholar] [CrossRef]
- Zhou, L.J.; Mo, Y.B.; Bu, X.; Wang, J.J.; Bai, J.; Zhang, J.W.; Cheng, A.B.; Ma, J.H.; Wang, Y.W.; Xie, Y.X. Erinacine Facilitates the Opening of the Mitochondrial Permeability Transition Pore Through the Inhibition of the PI3K/Akt/GSK-3beta Signaling Pathway in Human Hepatocellular Carcinoma. Cell Physiol. Biochem. 2018, 50, 851–867. [Google Scholar] [CrossRef]
- Yu, L.M.; Dong, X.; Xue, X.D.; Xu, S.; Zhang, X.; Xu, Y.L.; Wang, Z.S.; Wang, Y.; Gao, H.; Liang, Y.X.; et al. Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: Role of SIRT6. J. Pineal Res. 2021, 70, e12698. [Google Scholar] [CrossRef]
- Liu, H.; Peng, H.; Chen, S.; Liu, Y.; Xiang, H.; Chen, R.; Chen, W.; Zhao, S.; Chen, P.; Lu, H. S1PR2 antagonist protects endothelial cells against high glucose-induced mitochondrial apoptosis through the Akt/GSK-3beta signaling pathway. Biochem. Biophys. Res. Commun. 2017, 490, 1119–1124. [Google Scholar] [CrossRef]
- Al-Damry, N.T.; Attia, H.A.; Al-Rasheed, N.M.; Al-Rasheed, N.M.; Mohamad, R.A.; Al-Amin, M.A.; Dizmiri, N.; Atteya, M. Sitagliptin attenuates myocardial apoptosis via activating LKB-1/AMPK/Akt pathway and suppressing the activity of GSK-3beta and p38alpha/MAPK in a rat model of diabetic cardiomyopathy. Biomed. Pharmacother. 2018, 107, 347–358. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Zhang, J.; Gao, S.; Xu, T.; Yin, Y. JAK/STAT signaling in diabetic kidney disease. Front. Cell Dev. Biol. 2023, 11, 1233259. [Google Scholar] [CrossRef]
- Berthier, C.C.; Zhang, H.; Schin, M.; Henger, A.; Nelson, R.G.; Yee, B.; Boucherot, A.; Neusser, M.A.; Cohen, C.D.; Carter-Su, C.; et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009, 58, 469–477. [Google Scholar] [CrossRef]
- Huang, J.S.; Chuang, L.Y.; Guh, J.Y.; Chen, C.J.; Yang, Y.L.; Chiang, T.A.; Hung, M.Y.; Liao, T.N. Effect of nitric oxide-cGMP-dependent protein kinase activation on advanced glycation end-product-induced proliferation in renal fibroblasts. J. Am. Soc. Nephrol. 2005, 16, 2318–2329. [Google Scholar] [CrossRef]
- Nakajima, H.; Takenaka, M.; Kaimori, J.Y.; Hamano, T.; Iwatani, H.; Sugaya, T.; Ito, T.; Hori, M.; Imai, E. Activation of the signal transducer and activator of transcription signaling pathway in renal proximal tubular cells by albumin. J. Am. Soc. Nephrol. 2004, 15, 276–285. [Google Scholar] [CrossRef]
- Nightingale, J.; Patel, S.; Suzuki, N.; Buxton, R.; Takagi, K.I.; Suzuki, J.; Sumi, Y.; Imaizumi, A.; Mason, R.M.; Zhang, Z. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J. Am. Soc. Nephrol. 2004, 15, 21–32. [Google Scholar] [CrossRef]
- Banes, A.K.; Shaw, S.; Jenkins, J.; Redd, H.; Amiri, F.; Pollock, D.M.; Marrero, M.B. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am. J. Physiol. Renal Physiol. 2004, 286, F653–F659. [Google Scholar] [CrossRef]
- Wang, X.; Shaw, S.; Amiri, F.; Eaton, D.C.; Marrero, M.B. Inhibition of the Jak/STAT signaling pathway prevents the high glucose-induced increase in tgf-beta and fibronectin synthesis in mesangial cells. Diabetes 2002, 51, 3505–3509. [Google Scholar] [CrossRef]
- Zheng, C.; Huang, L.; Luo, W.; Yu, W.; Hu, X.; Guan, X.; Cai, Y.; Zou, C.; Yin, H.; Xu, Z.; et al. Inhibition of STAT3 in tubular epithelial cells prevents kidney fibrosis and nephropathy in STZ-induced diabetic mice. Cell Death Dis. 2019, 10, 848. [Google Scholar] [CrossRef]
- Hu, C.; Sun, L.; Xiao, L.; Han, Y.; Fu, X.; Xiong, X.; Xu, X.; Liu, Y.; Yang, S.; Liu, F.; et al. Insights into the Mechanisms Involved in the Expression and Regulation of Extracellular Matrix Proteins in Diabetic Nephropathy. Curr. Med. Chem. 2015, 22, 2858–2870. [Google Scholar] [CrossRef]
- Tian, H.; Yang, J.; Xie, Z.; Liu, J. Gliquidone Alleviates Diabetic Nephropathy by Inhibiting Notch/Snail Signaling Pathway. Cell Physiol. Biochem. 2018, 51, 2085–2097. [Google Scholar] [CrossRef]
- Nishad, R.; Mukhi, D.; Tahaseen, S.V.; Mungamuri, S.K.; Pasupulati, A.K. Growth hormone induces Notch1 signaling in podocytes and contributes to proteinuria in diabetic nephropathy. J. Biol. Chem. 2019, 294, 16109–16122. [Google Scholar] [CrossRef]
- Jing, Z.; Hu, L.; Su, Y.; Ying, G.; Ma, C.; Wei, J. Potential signaling pathway through which Notch regulates oxidative damage and apoptosis in renal tubular epithelial cells induced by high glucose. J. Recept. Signal Transduct. Res. 2021, 41, 357–362. [Google Scholar] [CrossRef]
- Vodosek Hojs, N.; Bevc, S.; Ekart, R.; Piko, N.; Petreski, T.; Hojs, R. Mineralocorticoid Receptor Antagonists in Diabetic Kidney Disease. Pharmaceuticals 2021, 14, 561. [Google Scholar] [CrossRef]
- Mende, C.W.; Samarakoon, R.; Higgins, P.J. Mineralocorticoid Receptor-Associated Mechanisms in Diabetic Kidney Disease and Clinical Significance of Mineralocorticoid Receptor Antagonists. Am. J. Nephrol. 2023, 54, 50–61. [Google Scholar] [CrossRef]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The multifaceted mineralocorticoid receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Cheung, A.K. Mineralocorticoid Receptor Antagonists in ESKD. Clin. J. Am. Soc. Nephrol. 2020, 15, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Le Tallec, L.; Lombes, M. The mineralocorticoid receptor: A journey exploring its diversity and specificity of action. Mol. Endocrinol. 2005, 19, 2211–2221. [Google Scholar] [CrossRef]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Hirohama, D.; Nishimoto, M.; Ayuzawa, N.; Kawarazaki, W.; Fujii, W.; Oba, S.; Shibata, S.; Marumo, T.; Fujita, T. Activation of Rac1-Mineralocorticoid Receptor Pathway Contributes to Renal Injury in Salt-Loaded db/db Mice. Hypertension 2021, 78, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Kadoya, H.; Satoh, M.; Sasaki, T.; Taniguchi, S.; Takahashi, M.; Kashihara, N. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J. 2015, 29, 3899–3910. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Conley, S.M.; Li, G.; Yuan, X.; Li, P.L. Rac1 GTPase Inhibition Blocked Podocyte Injury and Glomerular Sclerosis during Hyperhomocysteinemia via Suppression of Nucleotide-Binding Oligomerization Domain-Like Receptor Containing Pyrin Domain 3 Inflammasome Activation. Kidney Blood Press. Res. 2019, 44, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 705838. [Google Scholar] [CrossRef]
- Senger, D.R.; Wirth, D.F.; Hynes, R.O. Transformed mammalian cells secrete specific proteins and phosphoproteins. Cell 1979, 16, 885–893. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, Y.; Scherer, N.; Grundner-Culemann, F.; Lehtimaki, T.; Mishra, B.H.; Raitakari, O.T.; Nauck, M.; Eckardt, K.U.; Sekula, P.; et al. Genetics of osteopontin in patients with chronic kidney disease: The German Chronic Kidney Disease study. PLoS Genet. 2022, 18, e1010139. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Sakatsume, M.; Nishi, S.; Narita, I.; Arakawa, M.; Gejyo, F. Expression, roles, receptors, and regulation of osteopontin in the kidney. Kidney Int. 2001, 60, 1645–1657. [Google Scholar] [CrossRef]
- Sinha, S.K.; Mellody, M.; Carpio, M.B.; Damoiseaux, R.; Nicholas, S.B. Osteopontin as a Biomarker in Chronic Kidney Disease. Biomedicines 2023, 11, 1356. [Google Scholar] [CrossRef]
- Kelly, D.J.; Wilkinson-Berka, J.L.; Ricardo, S.D.; Cox, A.J.; Gilbert, R.E. Progression of tubulointerstitial injury by osteopontin-induced macrophage recruitment in advanced diabetic nephropathy of transgenic (mRen-2)27 rats. Nephrol. Dial. Transplant. 2002, 17, 985–991. [Google Scholar] [CrossRef]
- Susztak, K.; Bottinger, E.; Novetsky, A.; Liang, D.; Zhu, Y.; Ciccone, E.; Wu, D.; Dunn, S.; McCue, P.; Sharma, K. Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes 2004, 53, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, I.; Sekula, P.; Kotsis, F.; von Cube, M.; Cheng, Y.; Nadal, J.; Schmid, M.; Schneider, M.P.; Krane, V.; Nauck, M.; et al. Association of osteopontin with kidney function and kidney failure in chronic kidney disease patients: The GCKD study. Nephrol. Dial. Transplant. 2023, 38, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.; Nielsen, N.R.; Sorensen, M.R.; Jacobsen, L.N.; Ostenfeld, M.S.; Sorensen, E.S. Naturally Occurring N-Terminal Fragments of Bovine Milk Osteopontin Are Transported across Models of the Intestinal Barrier. Biomedicines 2023, 11, 893. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.I.; Bostrom, M.; Daeihagh, P.; Bowden, D.W. Genetic factors in diabetic nephropathy. Clin. J. Am. Soc. Nephrol. 2007, 2, 1306–1316. [Google Scholar] [CrossRef]
- Murea, M.; Ma, L.; Freedman, B.I. Genetic and environmental factors associated with type 2 diabetes and diabetic vascular complications. Rev. Diabet. Stud. 2012, 9, 6–22. [Google Scholar] [CrossRef]
- Thomas, M.C.; Groop, P.H.; Tryggvason, K. Towards understanding the inherited susceptibility for nephropathy in diabetes. Curr. Opin. Nephrol. Hypertens. 2012, 21, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Diabetic nephropathy-emerging epigenetic mechanisms. Nat. Rev. Nephrol. 2014, 10, 517–530. [Google Scholar] [CrossRef]
- Gu, H.F. Genetic and Epigenetic Studies in Diabetic Kidney Disease. Front. Genet. 2019, 10, 507. [Google Scholar] [CrossRef]
- Gu, H.F.; Brismar, K. Genetic association studies in diabetic nephropathy. Curr. Diabetes Rev. 2012, 8, 336–344. [Google Scholar] [CrossRef]
- Florez, J.C. Genetics of Diabetic Kidney Disease. Semin. Nephrol. 2016, 36, 474–480. [Google Scholar] [CrossRef]
- Moczulski, D.K.; Rogus, J.J.; Antonellis, A.; Warram, J.H.; Krolewski, A.S. Major susceptibility locus for nephropathy in type 1 diabetes on chromosome 3q: Results of novel discordant sib-pair analysis. Diabetes 1998, 47, 1164–1169. [Google Scholar] [CrossRef]
- Vionnet, N.; Tregouet, D.; Kazeem, G.; Gut, I.; Groop, P.H.; Tarnow, L.; Parving, H.H.; Hadjadj, S.; Forsblom, C.; Farrall, M.; et al. Analysis of 14 candidate genes for diabetic nephropathy on chromosome 3q in European populations: Strongest evidence for association with a variant in the promoter region of the adiponectin gene. Diabetes 2006, 55, 3166–3174. [Google Scholar] [CrossRef]
- He, B.; Osterholm, A.M.; Ojala, J.R.; Andersson, A.C.; Tryggvason, K. A remote cis-acting variant at 3q links glomerular NCK1 to diabetic nephropathy. PLoS ONE 2013, 8, e56414. [Google Scholar] [CrossRef]
- Janssen, B.; Hohenadel, D.; Brinkkoetter, P.; Peters, V.; Rind, N.; Fischer, C.; Rychlik, I.; Cerna, M.; Romzova, M.; de Heer, E.; et al. Carnosine as a protective factor in diabetic nephropathy: Association with a leucine repeat of the carnosinase gene CNDP1. Diabetes 2005, 54, 2320–2327. [Google Scholar] [CrossRef]
- Salem, R.M.; Todd, J.N.; Sandholm, N.; Cole, J.B.; Chen, W.M.; Andrews, D.; Pezzolesi, M.G.; McKeigue, P.M.; Hiraki, L.T.; Qiu, C.; et al. Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen. J. Am. Soc. Nephrol. 2019, 30, 2000–2016. [Google Scholar] [CrossRef]
- Khattab, A.; Torkamani, A. Nidogen-1 could play a role in diabetic kidney disease development in type 2 diabetes: A genome-wide association meta-analysis. Hum. Genom. 2022, 16, 47. [Google Scholar] [CrossRef]
- Jin, H.; Kim, Y.A.; Lee, Y.; Kwon, S.H.; Do, A.R.; Seo, S.; Won, S.; Seo, J.H. Identification of genetic variants associated with diabetic kidney disease in multiple Korean cohorts via a genome-wide association study mega-analysis. BMC Med. 2023, 21, 16. [Google Scholar] [CrossRef] [PubMed]
- Igo, R.P., Jr.; Iyengar, S.K.; Nicholas, S.B.; Goddard, K.A.; Langefeld, C.D.; Hanson, R.L.; Duggirala, R.; Divers, J.; Abboud, H.; Adler, S.G.; et al. Genomewide linkage scan for diabetic renal failure and albuminuria: The FIND study. Am. J. Nephrol. 2011, 33, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.K.; Sedor, J.R.; Freedman, B.I.; Kao, W.H.; Kretzler, M.; Keller, B.J.; Abboud, H.E.; Adler, S.G.; Best, L.G.; Bowden, D.W.; et al. Genome-Wide Association and Trans-ethnic Meta-Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet. 2015, 11, e1005352. [Google Scholar] [CrossRef] [PubMed]
- Thameem, F.; Igo, R.P., Jr.; Freedman, B.I.; Langefeld, C.; Hanson, R.L.; Schelling, J.R.; Elston, R.C.; Duggirala, R.; Nicholas, S.B.; Goddard, K.A.; et al. A genome-wide search for linkage of estimated glomerular filtration rate (eGFR) in the Family Investigation of Nephropathy and Diabetes (FIND). PLoS ONE 2013, 8, e81888. [Google Scholar] [CrossRef]
- Malhotra, A.; Igo, R.P., Jr.; Thameem, F.; Kao, W.H.; Abboud, H.E.; Adler, S.G.; Arar, N.H.; Bowden, D.W.; Duggirala, R.; Freedman, B.I.; et al. Genome-wide linkage scans for type 2 diabetes mellitus in four ethnically diverse populations-significant evidence for linkage on chromosome 4q in African Americans: The Family Investigation of Nephropathy and Diabetes Research Group. Diabetes Metab. Res. Rev. 2009, 25, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Schelling, J.R.; Abboud, H.E.; Nicholas, S.B.; Pahl, M.V.; Sedor, J.R.; Adler, S.G.; Arar, N.H.; Bowden, D.W.; Elston, R.C.; Freedman, B.I.; et al. Genome-wide scan for estimated glomerular filtration rate in multi-ethnic diabetic populations: The Family Investigation of Nephropathy and Diabetes (FIND). Diabetes 2008, 57, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.K.; Abboud, H.E.; Goddard, K.A.; Saad, M.F.; Adler, S.G.; Arar, N.H.; Bowden, D.W.; Duggirala, R.; Elston, R.C.; Hanson, R.L.; et al. Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: The family investigation of nephropathy and diabetes (FIND). Diabetes 2007, 56, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C. Epigenetic Mechanisms in Diabetic Kidney Disease. Curr. Diab. Rep. 2016, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.T.; van Diepen, J.A.; Riksen, N.P.; El-Osta, A. Epigenetics in diabetic nephropathy, immunity and metabolism. Diabetologia 2018, 61, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Qiu, C.; Liu, H.; Gluck, C.; Hsu, J.Y.; He, J.; Hsu, C.Y.; Sha, D.; Weir, M.R.; Isakova, T.; et al. Systematic integrated analysis of genetic and epigenetic variation in diabetic kidney disease. Proc. Natl. Acad. Sci. USA 2020, 117, 29013–29024. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.Y.; Tin, A.; Schlosser, P.; Ko, Y.A.; Qiu, C.; Yao, C.; Joehanes, R.; Grams, M.E.; Liang, L.; Gluck, C.A.; et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 2017, 8, 1286. [Google Scholar] [CrossRef]
- VanderJagt, T.A.; Neugebauer, M.H.; Morgan, M.; Bowden, D.W.; Shah, V.O. Epigenetic profiles of pre-diabetes transitioning to type 2 diabetes and nephropathy. World J. Diabetes 2015, 6, 1113–1121. [Google Scholar] [CrossRef]
- Marumo, T.; Yagi, S.; Kawarazaki, W.; Nishimoto, M.; Ayuzawa, N.; Watanabe, A.; Ueda, K.; Hirahashi, J.; Hishikawa, K.; Sakurai, H.; et al. Diabetes Induces Aberrant DNA Methylation in the Proximal Tubules of the Kidney. J. Am. Soc. Nephrol. 2015, 26, 2388–2397. [Google Scholar] [CrossRef]
- Aso, Y.; Yoshida, N.; Okumura, K.; Wakabayashi, S.; Matsutomo, R.; Takebayashi, K.; Inukai, T. Coagulation and inflammation in overt diabetic nephropathy: Association with hyperhomocysteinemia. Clin. Chim. Acta 2004, 348, 139–145. [Google Scholar] [CrossRef]
- Sayanthooran, S.; Magana-Arachchi, D.N.; Gunerathne, L.; Abeysekera, T. Potential diagnostic biomarkers for chronic kidney disease of unknown etiology (CKDu) in Sri Lanka: A pilot study. BMC Nephrol. 2017, 18, 31. [Google Scholar] [CrossRef]
- Aldemir, O.; Turgut, F.; Gokce, C. The association between methylation levels of targeted genes and albuminuria in patients with early diabetic kidney disease. Ren. Fail. 2017, 39, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Cao, R.F.; Yu, Y.; Sui, M.; Zhang, T.; Xu, J.Y.; Wang, X.M. A study on the correlation between MTHFR promoter methylation and diabetic nephropathy. Am. J. Transl. Res. 2016, 8, 4960–4967. [Google Scholar] [PubMed]
- Zhang, H.; Cai, X.; Yi, B.; Huang, J.; Wang, J.; Sun, J. Correlation of CTGF gene promoter methylation with CTGF expression in type 2 diabetes mellitus with or without nephropathy. Mol. Med. Rep. 2014, 9, 2138–2144. [Google Scholar] [CrossRef] [PubMed]
- Tampe, B.; Tampe, D.; Muller, C.A.; Sugimoto, H.; LeBleu, V.; Xu, X.; Muller, G.A.; Zeisberg, E.M.; Kalluri, R.; Zeisberg, M. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J. Am. Soc. Nephrol. 2014, 25, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Kourtidou, C.; Tziomalos, K. The Role of Histone Modifications in the Pathogenesis of Diabetic Kidney Disease. Int. J. Mol. Sci. 2023, 24, 6007. [Google Scholar] [CrossRef]
- Kuo, F.C.; Chao, C.T.; Lin, S.H. The Dynamics and Plasticity of Epigenetics in Diabetic Kidney Disease: Therapeutic Applications Vis-a-Vis. Int. J. Mol. Sci. 2022, 23, 843. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 2004, 279, 18091–18097. [Google Scholar] [CrossRef]
- Miao, F.; Wu, X.; Zhang, L.; Yuan, Y.C.; Riggs, A.D.; Natarajan, R. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J. Biol. Chem. 2007, 282, 13854–13863. [Google Scholar] [CrossRef]
- Miao, F.; Smith, D.D.; Zhang, L.; Min, A.; Feng, W.; Natarajan, R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: An epigenetic study in diabetes. Diabetes 2008, 57, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- Yuan, H.; Reddy, M.A.; Deshpande, S.; Jia, Y.; Park, J.T.; Lanting, L.L.; Jin, W.; Kato, M.; Xu, Z.G.; Das, S.; et al. Epigenetic Histone Modifications Involved in Profibrotic Gene Regulation by 12/15-Lipoxygenase and Its Oxidized Lipid Products in Diabetic Nephropathy. Antioxid. Redox Signal. 2016, 24, 361–375. [Google Scholar] [CrossRef]
- Kato, M.; Dang, V.; Wang, M.; Park, J.T.; Deshpande, S.; Kadam, S.; Mardiros, A.; Zhan, Y.; Oettgen, P.; Putta, S.; et al. TGF-beta induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci. Signal. 2013, 6, ra43. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.; Gao, C.; Chen, E.; Lightle, A.R.; Foulke, L.; Zhao, B.; Higgins, P.J.; Zhang, W. Loss of Histone H3 K79 Methyltransferase Dot1l Facilitates Kidney Fibrosis by Upregulating Endothelin 1 through Histone Deacetylase 2. J. Am. Soc. Nephrol. 2020, 31, 337–349. [Google Scholar] [CrossRef]
- Mimura, I. Epigenetic memory in kidney diseases. Kidney Int. 2016, 89, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Huang, Y.; Zhu, X.; Liu, C.; Yuan, Y.; Su, H.; Zhang, C.; Liu, C.; Xiong, M.; Qu, Y.; et al. Histone demethylase UTX is a therapeutic target for diabetic kidney disease. J. Physiol. 2019, 597, 1643–1660. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Hsu, Y.C.; Huang, Y.T.; Shih, Y.H.; Wang, C.J.; Chiang, W.C.; Chang, P.J. A KDM6A-KLF10 reinforcing feedback mechanism aggravates diabetic podocyte dysfunction. EMBO Mol. Med. 2019, 11, e9828. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Xiong, X.; Tang, B.; Ji, T.; Li, X.; Qu, X.; Li, W. hsa-miR-199b-3p Prevents the Epithelial-Mesenchymal Transition and Dysfunction of the Renal Tubule by Regulating E-cadherin through Targeting KDM6A in Diabetic Nephropathy. Oxid. Med. Cell Longev. 2021, 2021, 8814163. [Google Scholar] [CrossRef]
- Majumder, S.; Thieme, K.; Batchu, S.N.; Alghamdi, T.A.; Bowskill, B.B.; Kabir, M.G.; Liu, Y.; Advani, S.L.; White, K.E.; Geldenhuys, L.; et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J. Clin. Investig. 2018, 128, 483–499. [Google Scholar] [CrossRef]
- Cai, M.; Bompada, P.; Atac, D.; Laakso, M.; Groop, L.; De Marinis, Y. Epigenetic regulation of glucose-stimulated osteopontin (OPN) expression in diabetic kidney. Biochem. Biophys. Res. Commun. 2016, 469, 108–113. [Google Scholar] [CrossRef]
- Nathan, D.M.; Delahanty, L. Your health in the 21st century. The best ways to beat diabetes. Newsweek 2005, 145, 30–31. [Google Scholar]
- Schrauben, S.J.; Shou, H.; Zhang, X.; Anderson, A.H.; Bonventre, J.V.; Chen, J.; Coca, S.; Furth, S.L.; Greenberg, J.H.; Gutierrez, O.M.; et al. Association of Multiple Plasma Biomarker Concentrations with Progression of Prevalent Diabetic Kidney Disease: Findings from the Chronic Renal Insufficiency Cohort (CRIC) Study. J. Am. Soc. Nephrol. 2021, 32, 115–126. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, J.A.; Tatsch, E.; Hausen, B.S.; Bollick, Y.S.; Moretto, M.B.; Duarte, T.; Duarte, M.M.; Londero, S.W.; Premaor, M.O.; Comim, F.V.; et al. Urinary kidney injury molecule-1 and neutrophil gelatinase-associated lipocalin as indicators of tubular damage in normoalbuminuric patients with type 2 diabetes. Clin. Biochem. 2016, 49, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Gohda, T.; Tomino, Y. Novel biomarkers for the progression of diabetic nephropathy: Soluble TNF receptors. Curr. Diab. Rep. 2013, 13, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Yuruk Yildirim, Z.; Nayir, A.; Yilmaz, A.; Gedikbasi, A.; Bundak, R. Neutrophil Gelatinase-Associated Lipocalin as an Early Sign of Diabetic Kidney Injury in Children. J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Lacquaniti, A.; Donato, V.; Pintaudi, B.; Di Vieste, G.; Chirico, V.; Buemi, A.; Di Benedetto, A.; Arena, A.; Buemi, M. “Normoalbuminuric” diabetic nephropathy: Tubular damage and NGAL. Acta Diabetol. 2013, 50, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.E.; Sugaya, T.; Hovind, P.; Baba, T.; Parving, H.H.; Rossing, P. Urinary liver-type fatty acid-binding protein predicts progression to nephropathy in type 1 diabetic patients. Diabetes Care 2010, 33, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Panduru, N.M.; Forsblom, C.; Saraheimo, M.; Thorn, L.; Bierhaus, A.; Humpert, P.M.; Groop, P.H.; FinnDiane Study, G. Urinary liver-type fatty acid-binding protein and progression of diabetic nephropathy in type 1 diabetes. Diabetes Care 2013, 36, 2077–2083. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.D. The emerging role of angiotensin-converting enzyme-2 in the kidney. Curr. Opin. Nephrol. Hypertens. 2007, 16, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.J.; Wysocki, J.; Ye, M.; Lloveras, J.; Kanwar, Y.; Batlle, D. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007, 72, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Ambade, V.; Sing, P.; Somani, B.L.; Basanna, D. Urinary N-acetyl beta glucosaminidase and gamma glutamyl transferase as early markers of diabetic nephropathy. Indian. J. Clin. Biochem. 2006, 21, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.Y.; Hughes, K.; Chia, K.S.; Ng, V.; Ling, S.L. Urinary alpha1-microglobulin as a marker of nephropathy in type 2 diabetic Asian subjects in Singapore. Diabetes Care 2003, 26, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Petrica, L.; Vlad, A.; Gluhovschi, G.; Gadalean, F.; Dumitrascu, V.; Gluhovschi, C.; Velciov, S.; Bob, F.; Vlad, D.; Popescu, R.; et al. Proximal tubule dysfunction is associated with podocyte damage biomarkers nephrin and vascular endothelial growth factor in type 2 diabetes mellitus patients: A cross-sectional study. PLoS ONE 2014, 9, e112538. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutierrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef]
- Akimoto, T.; Yoshizawa, H.; Watanabe, Y.; Numata, A.; Yamazaki, T.; Takeshima, E.; Iwazu, K.; Komada, T.; Otani, N.; Morishita, Y.; et al. Characteristics of urinary and serum soluble Klotho protein in patients with different degrees of chronic kidney disease. BMC Nephrol. 2012, 13, 155. [Google Scholar] [CrossRef]
- Gutwein, P.; Schramme, A.; Abdel-Bakky, M.S.; Doberstein, K.; Hauser, I.A.; Ludwig, A.; Altevogt, P.; Gauer, S.; Hillmann, A.; Weide, T.; et al. ADAM10 is expressed in human podocytes and found in urinary vesicles of patients with glomerular kidney diseases. J. Biomed. Sci. 2010, 17, 3. [Google Scholar] [CrossRef]
- Cohen-Bucay, A.; Viswanathan, G. Urinary markers of glomerular injury in diabetic nephropathy. Int. J. Nephrol. 2012, 2012, 146987. [Google Scholar] [CrossRef]
- Narita, T.; Sasaki, H.; Hosoba, M.; Miura, T.; Yoshioka, N.; Morii, T.; Shimotomai, T.; Koshimura, J.; Fujita, H.; Kakei, M.; et al. Parallel increase in urinary excretion rates of immunoglobulin G, ceruloplasmin, transferrin, and orosomucoid in normoalbuminuric type 2 diabetic patients. Diabetes Care 2004, 27, 1176–1181. [Google Scholar] [CrossRef]
- Al-Rubeaan, K.; Siddiqui, K.; Al-Ghonaim, M.A.; Youssef, A.M.; Al-Sharqawi, A.H.; AlNaqeb, D. Assessment of the diagnostic value of different biomarkers in relation to various stages of diabetic nephropathy in type 2 diabetic patients. Sci. Rep. 2017, 7, 2684. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Haneda, M.; Koya, D.; Isshiki, K.; Kume, S.; Sugimoto, T.; Kawai, H.; Nishio, Y.; Kashiwagi, A.; Uzu, T.; et al. Association between urinary type IV collagen level and deterioration of renal function in type 2 diabetic patients without overt proteinuria. Diabetes Care 2010, 33, 1805–1810. [Google Scholar] [CrossRef]
- Fiseha, T. Urinary biomarkers for early diabetic nephropathy in type 2 diabetic patients. Biomark. Res. 2015, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Assal, H.S.; Tawfeek, S.; Rasheed, E.A.; El-Lebedy, D.; Thabet, E.H. Serum cystatin C and tubular urinary enzymes as biomarkers of renal dysfunction in type 2 diabetes mellitus. Clin. Med. Insights Endocrinol. Diabetes 2013, 6, 7–13. [Google Scholar] [CrossRef]
- Uslu, S.; Efe, B.; Alatas, O.; Kebapci, N.; Colak, O.; Demirustu, C.; Yoruk, A. Serum cystatin C and urinary enzymes as screening markers of renal dysfunction in diabetic patients. J. Nephrol. 2005, 18, 559–567. [Google Scholar] [PubMed]
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018, 117, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Fung, W.W.; Chan, G.C.; Ng, J.K.; Chow, K.M.; Szeto, C.C. Urinary and Kidney Podocalyxin and Podocin Levels in Diabetic Kidney Disease: A Kidney Biopsy Study. Kidney Med. 2023, 5, 100569. [Google Scholar] [CrossRef]
- Cho, N.J.; Han, D.J.; Lee, J.H.; Jang, S.H.; Kang, J.S.; Gil, H.W.; Park, S.; Lee, E.Y. Soluble klotho as a marker of renal fibrosis and podocyte injuries in human kidneys. PLoS ONE 2018, 13, e0194617. [Google Scholar] [CrossRef]
- Petrica, L.; Ursoniu, S.; Gadalean, F.; Vlad, A.; Gluhovschi, G.; Dumitrascu, V.; Vlad, D.; Gluhovschi, C.; Velciov, S.; Bob, F.; et al. Urinary podocyte-associated mRNA levels correlate with proximal tubule dysfunction in early diabetic nephropathy of type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2017, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Limonte, C.P.; Valo, E.; Drel, V.; Natarajan, L.; Darshi, M.; Forsblom, C.; Henderson, C.M.; Hoofnagle, A.N.; Ju, W.; Kretzler, M.; et al. Urinary Proteomics Identifies Cathepsin D as a Biomarker of Rapid eGFR Decline in Type 1 Diabetes. Diabetes Care 2022, 45, 1416–1427. [Google Scholar] [CrossRef]
- Sharma, K.; Zhang, G.; Hansen, J.; Bjornstad, P.; Lee, H.J.; Menon, R.; Hejazi, L.; Liu, J.J.; Franzone, A.; Looker, H.C.; et al. Endogenous adenine mediates kidney injury in diabetic models and predicts diabetic kidney disease in patients. J. Clin. Investig. 2023, 133, e170341. [Google Scholar] [CrossRef] [PubMed]
- Campion, C.G.; Sanchez-Ferras, O.; Batchu, S.N. Potential Role of Serum and Urinary Biomarkers in Diagnosis and Prognosis of Diabetic Nephropathy. Can. J. Kidney Health Dis. 2017, 4, 2054358117705371. [Google Scholar] [CrossRef] [PubMed]
- Gohda, T.; Walker, W.H.; Wolkow, P.; Lee, J.E.; Warram, J.H.; Krolewski, A.S.; Niewczas, M.A. Elevated urinary excretion of immunoglobulins in nonproteinuric patients with type 1 diabetes. Am. J. Physiol. Renal Physiol. 2012, 303, F157–F162. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Ferri, C.M.; Sanchez-Quintana, F.; Perez-Castro, A.; Gonzalez-Luis, A.; Martin-Nunez, E.; Mora-Fernandez, C.; Navarro-Gonzalez, J.F. Inflammatory Cytokines in Diabetic Kidney Disease: Pathophysiologic and Therapeutic Implications. Front. Med. 2020, 7, 628289. [Google Scholar] [CrossRef]
- Hinokio, Y.; Suzuki, S.; Hirai, M.; Suzuki, C.; Suzuki, M.; Toyota, T. Urinary excretion of 8-oxo-7, 8-dihydro-2′-deoxyguanosine as a predictor of the development of diabetic nephropathy. Diabetologia 2002, 45, 877–882. [Google Scholar] [CrossRef]
- Machowska, A.; Sun, J.; Qureshi, A.R.; Isoyama, N.; Leurs, P.; Anderstam, B.; Heimburger, O.; Barany, P.; Stenvinkel, P.; Lindholm, B. Plasma Pentosidine and Its Association with Mortality in Patients with Chronic Kidney Disease. PLoS ONE 2016, 11, e0163826. [Google Scholar] [CrossRef]
- Mauer, M.; Doria, A. Uric Acid and Diabetic Nephropathy Risk. Contrib. Nephrol. 2018, 192, 103–109. [Google Scholar] [CrossRef]
- Vodosek Hojs, N.; Bevc, S.; Ekart, R.; Hojs, R. Oxidative Stress Markers in Chronic Kidney Disease with Emphasis on Diabetic Nephropathy. Antioxidants 2020, 9, 925. [Google Scholar] [CrossRef]
- Altuhafi, A.; Altun, M.; Hadwan, M.H. The Correlation between Selenium-Dependent Glutathione Peroxidase Activity and Oxidant/Antioxidant Balance in Sera of Diabetic Patients with Nephropathy. Rep. Biochem. Mol. Biol. 2021, 10, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Sheira, G.; Noreldin, N.; Tamer, A.; Saad, M. Urinary biomarker N-acetyl-beta-D-glucosaminidase can predict severity of renal damage in diabetic nephropathy. J. Diabetes Metab. Disord. 2015, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2020, 98, S1–S115. [CrossRef]
- KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [CrossRef] [PubMed]
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes Management in Chronic Kidney Disease: A Consensus Report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Diabetes Care 2022, 45, 3075–3090. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Critical Reanalysis of the Mechanisms Underlying the Cardiorenal Benefits of SGLT2 Inhibitors and Reaffirmation of the Nutrient Deprivation Signaling/Autophagy Hypothesis. Circulation 2022, 146, 1383–1405. [Google Scholar] [CrossRef]
- Packer, M. SGLT2 inhibitors: Role in protective reprogramming of cardiac nutrient transport and metabolism. Nat. Rev. Cardiol. 2023, 20, 443–462. [Google Scholar] [CrossRef]
- Turan, B.; Durak, A.; Olgar, Y.; Tuncay, E. Comparisons of pleiotropic effects of SGLT2 inhibition and GLP-1 agonism on cardiac glucose intolerance in heart dysfunction. Mol. Cell Biochem. 2022, 477, 2609–2625. [Google Scholar] [CrossRef]
- Palm, F.; Cederberg, J.; Hansell, P.; Liss, P.; Carlsson, P.O. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 2003, 46, 1153–1160. [Google Scholar] [CrossRef]
- Fine, L.G.; Norman, J.T. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: From hypothesis to novel therapeutics. Kidney Int. 2008, 74, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur. J. Pharmacol. 2013, 715, 246–255. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Panchapakesan, U.; Pegg, K.; Gross, S.; Komala, M.G.; Mudaliar, H.; Forbes, J.; Pollock, C.; Mather, A. Effects of SGLT2 inhibition in human kidney proximal tubular cells--renoprotection in diabetic nephropathy? PLoS ONE 2013, 8, e54442. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.A.; Ward, M.S.; Fotheringham, A.K.; Zhuang, A.; Borg, D.J.; Flemming, N.B.; Harvie, B.M.; Kinneally, T.L.; Yeh, S.M.; McCarthy, D.A.; et al. Once daily administration of the SGLT2 inhibitor, empagliflozin, attenuates markers of renal fibrosis without improving albuminuria in diabetic db/db mice. Sci. Rep. 2016, 6, 26428. [Google Scholar] [CrossRef] [PubMed]
- The Nuffield Department of Population Health Renal Studies Group; SGLT2 Inhibitor Meta-Analysis Cardio-Renal Trialists’ Consortium. Impact of diabetes on the effects of sodium glucose co-transporter-2 inhibitors on kidney outcomes: Collaborative meta-analysis of large placebo-controlled trials. Lancet 2022, 400, 1788–1801. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br. J. Pharmacol. 2022, 179, 3220–3234. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, S.B. Novel Anti-inflammatory and Anti-fibrotic Agents for Diabetic Kidney Disease-From Bench to Bedside. Adv. Chronic Kidney Dis. 2021, 28, 378–390. [Google Scholar] [CrossRef]
- Barton, M.; Yanagisawa, M. Endothelin: 20 years from discovery to therapy. Can. J. Physiol. Pharmacol. 2008, 86, 485–498. [Google Scholar] [CrossRef]
- Martinez-Diaz, I.; Martos, N.; Llorens-Cebria, C.; Alvarez, F.J.; Bedard, P.W.; Vergara, A.; Jacobs-Cacha, C.; Soler, M.J. Endothelin Receptor Antagonists in Kidney Disease. Int. J. Mol. Sci. 2023, 24, 3427. [Google Scholar] [CrossRef]
- Chung, E.Y.M.; Badve, S.V.; Heerspink, H.J.L.; Wong, M.G. Endothelin receptor antagonists in kidney protection for diabetic kidney disease and beyond? Nephrology 2023, 28, 97–108. [Google Scholar] [CrossRef]
- Dhaun, N.; Goddard, J.; Webb, D.J. The endothelin system and its antagonism in chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Milon, M.; Virsolvy, A.; Henique, C.; Schmitt, A.; Masse, J.M.; Kotelevtsev, Y.; Yanagisawa, M.; Webb, D.J.; Richard, S.; et al. Direct action of endothelin-1 on podocytes promotes diabetic glomerulosclerosis. J. Am. Soc. Nephrol. 2014, 25, 1050–1062. [Google Scholar] [CrossRef]
- Collino, F.; Bussolati, B.; Gerbaudo, E.; Marozio, L.; Pelissetto, S.; Benedetto, C.; Camussi, G. Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am. J. Physiol. Renal Physiol. 2008, 294, F1185–F1194. [Google Scholar] [CrossRef]
- Ebefors, K.; Wiener, R.J.; Yu, L.; Azeloglu, E.U.; Yi, Z.; Jia, F.; Zhang, W.; Baron, M.H.; He, J.C.; Haraldsson, B.; et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int. 2019, 96, 957–970. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Goddard, J.; Johnston, N.R.; Hand, M.F.; Cumming, A.D.; Rabelink, T.J.; Rankin, A.J.; Webb, D.J. Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: A comparison of selective and combined endothelin receptor blockade. Circulation 2004, 109, 1186–1193. [Google Scholar] [CrossRef]
- Ortmann, J.; Amann, K.; Brandes, R.P.; Kretzler, M.; Munter, K.; Parekh, N.; Traupe, T.; Lange, M.; Lattmann, T.; Barton, M. Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension 2004, 44, 974–981. [Google Scholar] [CrossRef]
- Kohan, D.E.; Barton, M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014, 86, 896–904. [Google Scholar] [CrossRef]
- Hocher, B.; Schwarz, A.; Reinbacher, D.; Jacobi, J.; Lun, A.; Priem, F.; Bauer, C.; Neumayer, H.H.; Raschack, M. Effects of endothelin receptor antagonists on the progression of diabetic nephropathy. Nephron 2001, 87, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Pollock, D.M. Endothelin antagonists for diabetic and non-diabetic chronic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 573–579. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Coll, B.; Andress, D.; Brennan, J.J.; Tang, H.; Houser, M.; Correa-Rotter, R.; Kohan, D.; Lambers Heerspink, H.J.; Makino, H.; et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1083–1093. [Google Scholar] [CrossRef]
- Wenzel, R.R.; Littke, T.; Kuranoff, S.; Jurgens, C.; Bruck, H.; Ritz, E.; Philipp, T.; Mitchell, A.; SPP301 (Avosentan) Endothelin Antagonist Evaluation in Diabetic Nephropathy Study Investigators. Avosentan reduces albumin excretion in diabetics with macroalbuminuria. J. Am. Soc. Nephrol. 2009, 20, 655–664. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Kohan, D.E.; de Zeeuw, D. New insights from SONAR indicate adding sodium glucose co-transporter 2 inhibitors to an endothelin receptor antagonist mitigates fluid retention and enhances albuminuria reduction. Kidney Int. 2021, 99, 346–349. [Google Scholar] [CrossRef]
- Sawaf, H.; Thomas, G.; Taliercio, J.J.; Nakhoul, G.; Vachharajani, T.J.; Mehdi, A. Therapeutic Advances in Diabetic Nephropathy. J. Clin. Med. 2022, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N. Engl. J. Med. 1993, 329, 1456–1462. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Neumiller, J.J.; Johnson, E.J.; Dieter, B.; Tuttle, K.R. Sodium-Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes 2019, 68, 248–257. [Google Scholar] [CrossRef]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Renal Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Tsogka, K.; Nikolaou, E.; Liakopoulos, V.; Stefanidis, I. A unifying model of glucotoxicity in human renal proximal tubular epithelial cells and the effect of the SGLT2 inhibitor dapagliflozin. Int. Urol. Nephrol. 2020, 52, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Li, J.; Neuen, B.L.; Neal, B.; Arnott, C.; Parikh, C.R.; Coca, S.G.; Perkovic, V.; Mahaffey, K.W.; Yavin, Y.; et al. Effects of the SGLT2 inhibitor canagliflozin on plasma biomarkers TNFR-1, TNFR-2 and KIM-1 in the CANVAS trial. Diabetologia 2021, 64, 2147–2158. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M.; et al. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: The FIDELITY pooled analysis. Eur. Heart J. 2022, 43, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Green, D.; Jamerson, K.; Ruilope, L.M.; Kuranoff, S.J.; Littke, T.; Viberti, G.; Group, A.S. Avosentan for overt diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 527–535. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Factors that promote diabetes and DKD. The diagram illustrates that the progression of DKD encompasses a range of factors, including metabolic, hemodynamic, inflammatory, fibrotic processes, and genetic and epigenetic factors. These components impact various pathways that oversee intricate intracellular signaling networks, ultimately resulting in both functional and structural alterations within the kidney. Abbreviations: DKD: diabetic kidney disease, MR: mineralocorticoid receptor, AGE: advanced glycation end products, ACE: angiotensin-converting enzyme, RAAS: renin–angiotensin–aldosterone system, TGF-β: transforming growth factor-β, MAPKs: mitogen-activated protein kinases, PI3K/AKT: phosphatidylinositol-3-kinase/Ak strain transforming, JAK/STAT: Janus kinase/signal transducers and activators of transcription.
Figure 1.
Factors that promote diabetes and DKD. The diagram illustrates that the progression of DKD encompasses a range of factors, including metabolic, hemodynamic, inflammatory, fibrotic processes, and genetic and epigenetic factors. These components impact various pathways that oversee intricate intracellular signaling networks, ultimately resulting in both functional and structural alterations within the kidney. Abbreviations: DKD: diabetic kidney disease, MR: mineralocorticoid receptor, AGE: advanced glycation end products, ACE: angiotensin-converting enzyme, RAAS: renin–angiotensin–aldosterone system, TGF-β: transforming growth factor-β, MAPKs: mitogen-activated protein kinases, PI3K/AKT: phosphatidylinositol-3-kinase/Ak strain transforming, JAK/STAT: Janus kinase/signal transducers and activators of transcription.

Figure 2.
Role of metabolic factors in the development of DKD. The diagram depicts the impact of metabolic disruptions in hyperglycemia on various pathways, namely PKC, AGE, hexosamine, and the polyol pathway. The PKC pathway is linked to heightened production of ECM, especially collagen IV, leading to the expansion of mesangial cells and the development of glomerulosclerosis. The interaction between AGE and RAGE triggers downstream signaling molecules, including MAPK, p38, SAPK/JNK, ERK1/2, and JAK/STAT. The hexosamine pathway is responsible for generating glucosamine-6-p, which in turn promotes the release of cytokines such as TGF-β, ICAM-1, VCAM-1, TNF-α, CTGF, and PAI-1. The polyol pathway results in a redox imbalance between NADH and NAD+ in diabetes mellitus. Collectively, these processes contribute to the progression of DKD. Abbreviations: DKD: diabetic kidney disease, glucosamine-6-P: glucosamine-6-phosphate, PKC: protein kinase C, AGE: advanced glycation end products, ECM: extracellular matrix, RAGE: receptor for advanced glycation end products, SAPK/JNK: stress-activated protein kinases/Jun amino-terminal kinases, ERK1/2: extracellular signal-regulated kinase 1/2, JAK/STAT: Janus kinase/signal transducers and activators of transcription, TGF-β: transforming growth factor- β, ICAM-1: intercellular adhesion molecule 1, VCAM-1: vascular cell adhesion molecule 1, TNF-α: tumor necrosis factor alpha, CTGF: connective tissue growth factor, PAI-1: plasminogen activator inhibitor 1.
Figure 2.
Role of metabolic factors in the development of DKD. The diagram depicts the impact of metabolic disruptions in hyperglycemia on various pathways, namely PKC, AGE, hexosamine, and the polyol pathway. The PKC pathway is linked to heightened production of ECM, especially collagen IV, leading to the expansion of mesangial cells and the development of glomerulosclerosis. The interaction between AGE and RAGE triggers downstream signaling molecules, including MAPK, p38, SAPK/JNK, ERK1/2, and JAK/STAT. The hexosamine pathway is responsible for generating glucosamine-6-p, which in turn promotes the release of cytokines such as TGF-β, ICAM-1, VCAM-1, TNF-α, CTGF, and PAI-1. The polyol pathway results in a redox imbalance between NADH and NAD+ in diabetes mellitus. Collectively, these processes contribute to the progression of DKD. Abbreviations: DKD: diabetic kidney disease, glucosamine-6-P: glucosamine-6-phosphate, PKC: protein kinase C, AGE: advanced glycation end products, ECM: extracellular matrix, RAGE: receptor for advanced glycation end products, SAPK/JNK: stress-activated protein kinases/Jun amino-terminal kinases, ERK1/2: extracellular signal-regulated kinase 1/2, JAK/STAT: Janus kinase/signal transducers and activators of transcription, TGF-β: transforming growth factor- β, ICAM-1: intercellular adhesion molecule 1, VCAM-1: vascular cell adhesion molecule 1, TNF-α: tumor necrosis factor alpha, CTGF: connective tissue growth factor, PAI-1: plasminogen activator inhibitor 1.

Figure 3.
Role of hemodynamic factors in the development of DKD. The figure demonstrates the process by which diabetes gives rise to hemodynamic changes, characterized by elevated systemic blood pressure and heightened intraglomerular pressure. These factors collectively encourage angiotensin II-induced vasoconstriction. The amplified glomerular capillary pressure and increased glomerular size led to a more permeable GBM, consequently resulting in albuminuria. These combined effects contribute to the development of DKD. Abbreviations: SGLT2: sodium–glucose cotransporter 2, ET: endothelin, DKD: diabetic kidney disease, GBM: glomerular basement membrane.
Figure 3.
Role of hemodynamic factors in the development of DKD. The figure demonstrates the process by which diabetes gives rise to hemodynamic changes, characterized by elevated systemic blood pressure and heightened intraglomerular pressure. These factors collectively encourage angiotensin II-induced vasoconstriction. The amplified glomerular capillary pressure and increased glomerular size led to a more permeable GBM, consequently resulting in albuminuria. These combined effects contribute to the development of DKD. Abbreviations: SGLT2: sodium–glucose cotransporter 2, ET: endothelin, DKD: diabetic kidney disease, GBM: glomerular basement membrane.

Figure 4.
Role of inflammatory factors in the development of DKD. The diagram illustrates how inflammation plays a role in the development of DKD. In diabetes, there is an infiltration and activation of immune cells, including macrophages, T-cells, and B-cells, within renal tissue. This leads to an increased expression of proinflammatory cytokines like IL-1, IL-6, IL-18, and TNF-α. Consequently, this intensifies the inflammatory responses within the renal tissue, contributing to its damage. Abbreviations: DKD: diabetic kidney disease, IL: interleukin, TNF-α: tumor necrosis factor alpha, MR: mineralocorticoid receptor, ROS: reactive oxygen species, Mφ: macrophage, DC: dendritic cell.
Figure 4.
Role of inflammatory factors in the development of DKD. The diagram illustrates how inflammation plays a role in the development of DKD. In diabetes, there is an infiltration and activation of immune cells, including macrophages, T-cells, and B-cells, within renal tissue. This leads to an increased expression of proinflammatory cytokines like IL-1, IL-6, IL-18, and TNF-α. Consequently, this intensifies the inflammatory responses within the renal tissue, contributing to its damage. Abbreviations: DKD: diabetic kidney disease, IL: interleukin, TNF-α: tumor necrosis factor alpha, MR: mineralocorticoid receptor, ROS: reactive oxygen species, Mφ: macrophage, DC: dendritic cell.

Figure 5.
Role of fibrotic factors in the development of DKD. The diagram depicts the involvement of renal fibrosis in the development of DKD. This fibrotic process encompasses various pathways, including TGF-β, MAPK, Wnt/β-catenin, PI3K/AKT, JAK/STAT, and NOTCH signaling. Within the TGF-β signaling pathway, Smad 2/3 mediates the production of ECM. The MAPK pathway leads to an increase in signaling transduction mediated by P38MAPK, ERK1/2, and JNK, resulting in elevated collagen IV levels. The Wnt/β-catenin pathway contributes to podocyte loss and mesangial cell apoptosis through the regulation of WT-1-associated genes and the cleavage of cas-3 and PARP, respectively. The collective action of the PI3K/AKT-JAK/STAT-NOTCH signaling pathways promotes PI3K/AKT, STAT1/3, and Snail signaling, leading to the induced expression of fibronectin/collagen IV, TGF-β1, VEGF, ACE, α-SMA, MMP2/9. Ultimately, these molecules drive the process of renal fibrosis in DKD. Abbreviations: DKD: diabetic kidney disease, WT-1: Wilms tumor-1, a master regulator of gene expression in podocytes, TGF-β: transforming growth factor-beta, MAPK: mitogen-activated protein kinases, ERK1/2: extracellular signal-regulated kinase 1/2, JNK: c-Jun amino-terminal kinase, Cas-3: caspase-3, PARP: polyADP-ribose polymerase, PI3K/AKT: phosphatidylinositol-3-kinase/Ak strain transforming, STAT1/3: signal transducer and activator of transcription 1/3, JAK/STAT: Janus kinase/signal transducers and activators of transcription, Smad 2/3: suppressor of mothers against decapentaplegic 2/3, ECM: extracellular matrix, VEGF: vascular endothelial growth factor, ACE: angiotensin-converting enzyme, a-SMA: alpha smooth muscle actin, MMP2/9: matrix metalloproteinase2/9.
Figure 5.
Role of fibrotic factors in the development of DKD. The diagram depicts the involvement of renal fibrosis in the development of DKD. This fibrotic process encompasses various pathways, including TGF-β, MAPK, Wnt/β-catenin, PI3K/AKT, JAK/STAT, and NOTCH signaling. Within the TGF-β signaling pathway, Smad 2/3 mediates the production of ECM. The MAPK pathway leads to an increase in signaling transduction mediated by P38MAPK, ERK1/2, and JNK, resulting in elevated collagen IV levels. The Wnt/β-catenin pathway contributes to podocyte loss and mesangial cell apoptosis through the regulation of WT-1-associated genes and the cleavage of cas-3 and PARP, respectively. The collective action of the PI3K/AKT-JAK/STAT-NOTCH signaling pathways promotes PI3K/AKT, STAT1/3, and Snail signaling, leading to the induced expression of fibronectin/collagen IV, TGF-β1, VEGF, ACE, α-SMA, MMP2/9. Ultimately, these molecules drive the process of renal fibrosis in DKD. Abbreviations: DKD: diabetic kidney disease, WT-1: Wilms tumor-1, a master regulator of gene expression in podocytes, TGF-β: transforming growth factor-beta, MAPK: mitogen-activated protein kinases, ERK1/2: extracellular signal-regulated kinase 1/2, JNK: c-Jun amino-terminal kinase, Cas-3: caspase-3, PARP: polyADP-ribose polymerase, PI3K/AKT: phosphatidylinositol-3-kinase/Ak strain transforming, STAT1/3: signal transducer and activator of transcription 1/3, JAK/STAT: Janus kinase/signal transducers and activators of transcription, Smad 2/3: suppressor of mothers against decapentaplegic 2/3, ECM: extracellular matrix, VEGF: vascular endothelial growth factor, ACE: angiotensin-converting enzyme, a-SMA: alpha smooth muscle actin, MMP2/9: matrix metalloproteinase2/9.

Figure 6.
Mineralocorticoid receptor (MR) and aldosterone in DKD. The diagram depicts the involvement of the MR and aldosterone in the development of DKD. The MR functions as an intracellular receptor, instigating the inflammatory cascade with aldosterone by generating ROS in the mitochondria, a process further amplified by Rac1. Both Rac1 and aldosterone contribute to fibrosis and inflammation by activating inflammasomes, NLRP3. The activation of the NLRP3 inflammasome, results in renal inflammation, fibrosis, and glomerular sclerosis. Abbreviations: DKD: diabetic kidney disease, MR: mineralocorticoid receptor, ROS: reactive oxygen species NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3.
Figure 6.
Mineralocorticoid receptor (MR) and aldosterone in DKD. The diagram depicts the involvement of the MR and aldosterone in the development of DKD. The MR functions as an intracellular receptor, instigating the inflammatory cascade with aldosterone by generating ROS in the mitochondria, a process further amplified by Rac1. Both Rac1 and aldosterone contribute to fibrosis and inflammation by activating inflammasomes, NLRP3. The activation of the NLRP3 inflammasome, results in renal inflammation, fibrosis, and glomerular sclerosis. Abbreviations: DKD: diabetic kidney disease, MR: mineralocorticoid receptor, ROS: reactive oxygen species NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3.

Figure 7.
OPN-mediated fibrosis in DKD. The diagram illustrates OPN-mediated signaling pathways. The detrimental impacts of OPN occur via its interaction with receptors, integrins, and CD44. Binding to these receptors leads to significant proinflammatory functions, enabling OPN to trigger the activation of various pathways, including cell survival, cell proliferation, angiogenesis, migration, and fibrosis. Abbreviations: OPN: osteopontin, ntOPN: N-terminal osteopontin, TGF-β: transforming growth factor-β, ERK/MAPK: extracellular signal-regulated kinase/mitogen-activated protein kinase, JNK/MAPK: jun N-terminal kinase/mitogen-activated protein kinase, PI3K/AKT/mTOR: phosphatidylinositol-3-kinase/Ak strain transforming/mammalian target of rapamycin, FAK/ERK1/2/NF-κB: focal adhesion kinase/extracellular signal-regulated kinase 1 and 2/nuclear factor kappa B.
Figure 7.
OPN-mediated fibrosis in DKD. The diagram illustrates OPN-mediated signaling pathways. The detrimental impacts of OPN occur via its interaction with receptors, integrins, and CD44. Binding to these receptors leads to significant proinflammatory functions, enabling OPN to trigger the activation of various pathways, including cell survival, cell proliferation, angiogenesis, migration, and fibrosis. Abbreviations: OPN: osteopontin, ntOPN: N-terminal osteopontin, TGF-β: transforming growth factor-β, ERK/MAPK: extracellular signal-regulated kinase/mitogen-activated protein kinase, JNK/MAPK: jun N-terminal kinase/mitogen-activated protein kinase, PI3K/AKT/mTOR: phosphatidylinositol-3-kinase/Ak strain transforming/mammalian target of rapamycin, FAK/ERK1/2/NF-κB: focal adhesion kinase/extracellular signal-regulated kinase 1 and 2/nuclear factor kappa B.

Figure 8.
Epigenetics in DKD. Histone modifications, specifically acetylation and methylation, along with DNA methylation, are linked to the aberrant regulation of genes associated with inflammation and fibrosis in DKD. Abbreviations: DM: diabetes mellitus, DKD: diabetic kidney disease, Me: methylation, Ac: acetylation, DNA: deoxyribonucleic acid.
Figure 8.
Epigenetics in DKD. Histone modifications, specifically acetylation and methylation, along with DNA methylation, are linked to the aberrant regulation of genes associated with inflammation and fibrosis in DKD. Abbreviations: DM: diabetes mellitus, DKD: diabetic kidney disease, Me: methylation, Ac: acetylation, DNA: deoxyribonucleic acid.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sinha, S.K.; Nicholas, S.B. Pathomechanisms of Diabetic Kidney Disease. J. Clin. Med. 2023, 12, 7349. https://doi.org/10.3390/jcm12237349
AMA Style
Sinha SK, Nicholas SB. Pathomechanisms of Diabetic Kidney Disease. Journal of Clinical Medicine. 2023; 12(23):7349. https://doi.org/10.3390/jcm12237349
Chicago/Turabian StyleSinha, Satyesh K., and Susanne B. Nicholas. 2023. "Pathomechanisms of Diabetic Kidney Disease" Journal of Clinical Medicine 12, no. 23: 7349. https://doi.org/10.3390/jcm12237349
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.