
Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments
 , , , ,
, , , , 
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
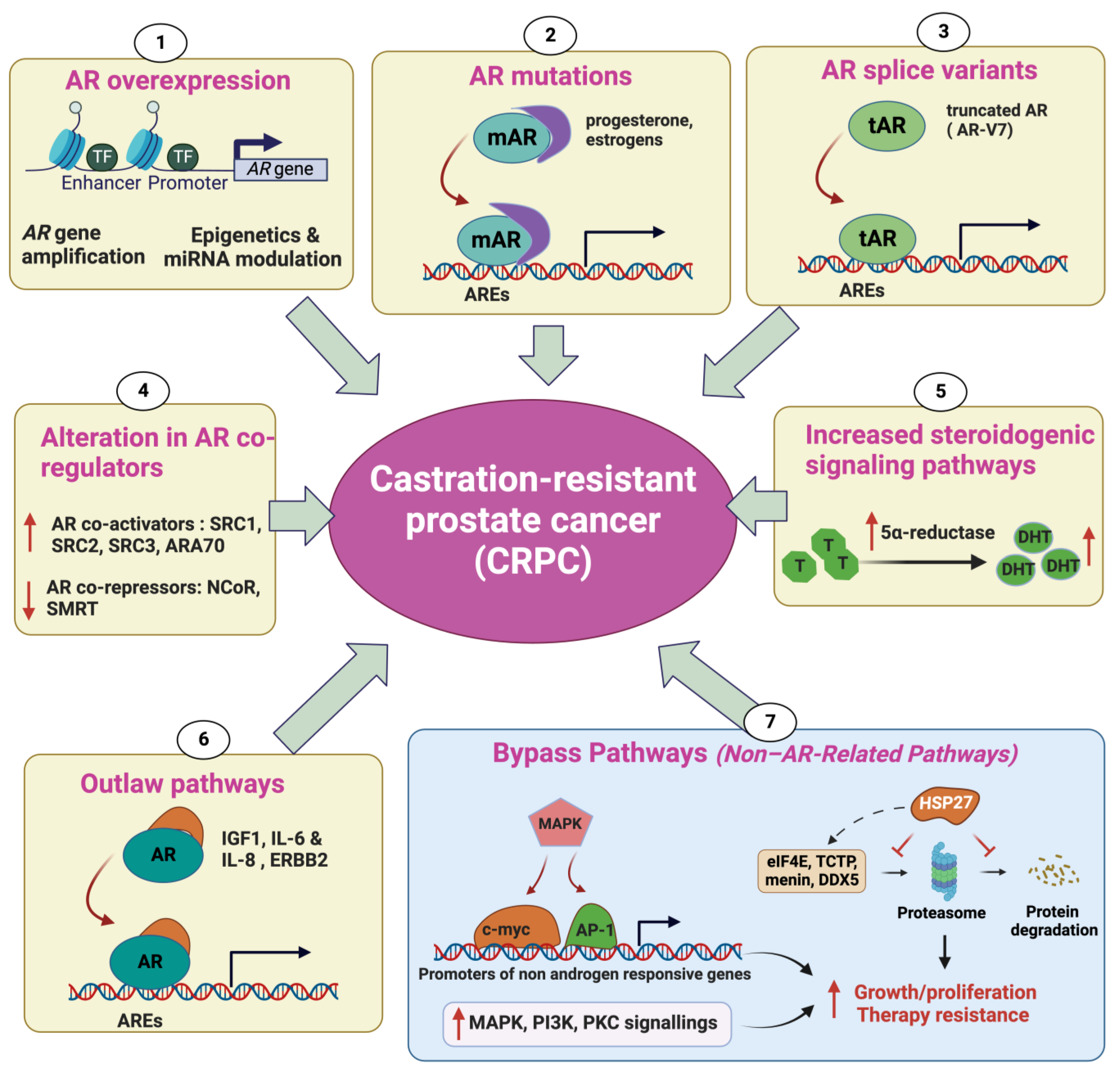
2. Mechanisms Underlying Castration-Resistant Prostate Cancer
2.1. Androgen Receptor Overexpression
2.2. AR Mutations
2.3. Alteration in AR CoRegulators
2.4. Increased Steroidogenic Signaling Pathways
2.5. Outlaw Pathways
2.6. Non-AR-Related Pathways: Bypass Pathways
2.7. Evolution of t-NEPC from CRPC
3. Treatment Options for CRPC
3.1. Androgen Deprivation Therapy (ADT)
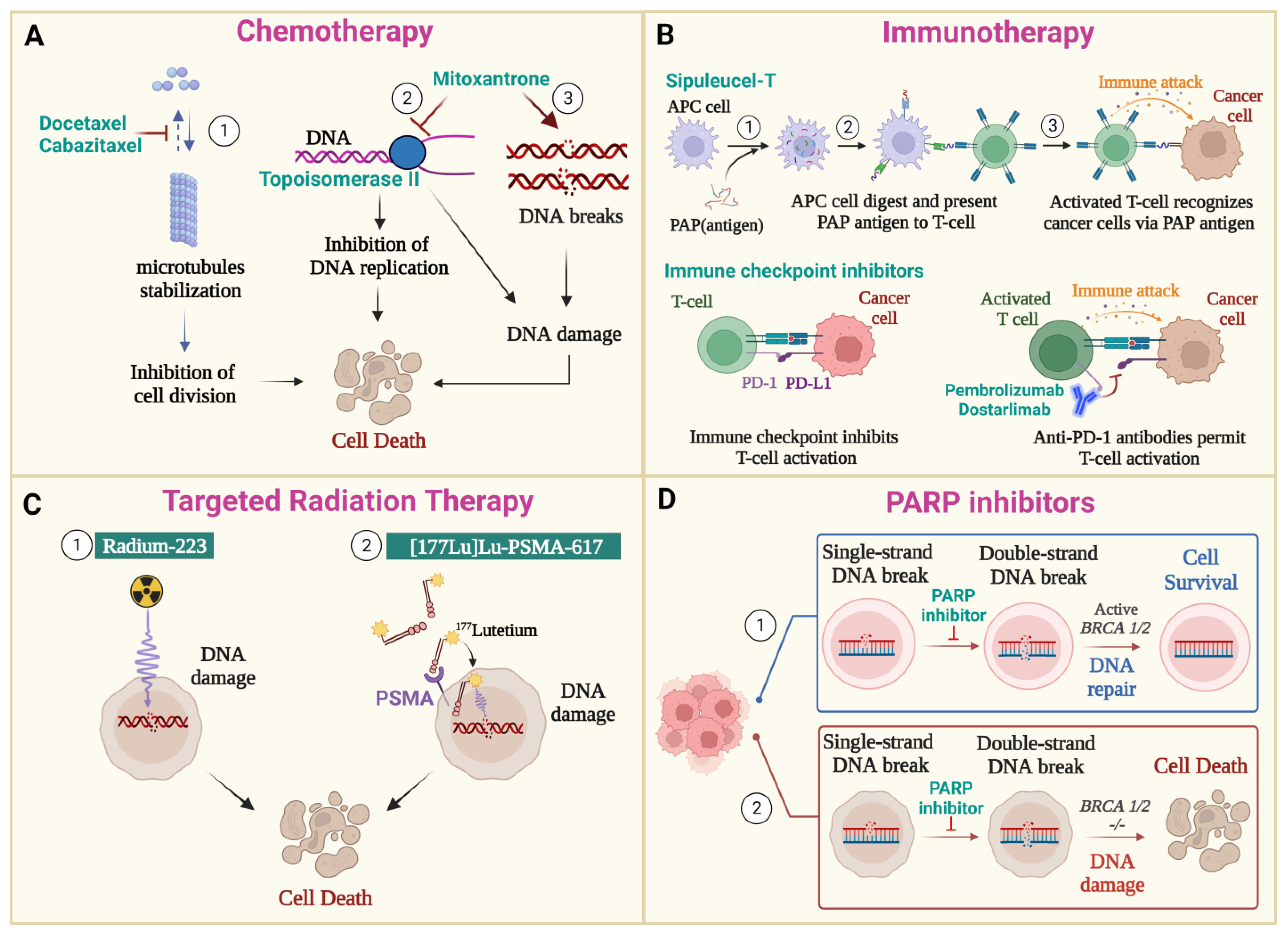
3.2. Chemotherapy
3.3. Immunotherapy
3.4. Radiation Therapy
3.4.1. PSMA-Targeting Radioligand Therapy
3.4.2. Radium-223 Dichloride
3.5. PARP Inhibitors
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA. Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Scher, H.I.; Morris, M.J.; Stadler, W.M.; Higano, C.S.; Halabi, S.; Smith, M.R.; Basch, E.M.; Fizazi, K.; Ryan, C.J.; Antonarakis, E.S.; et al. The Prostate Cancer Working Group 3 (PCWG3) Consensus for Trials in Castration-Resistant Prostate Cancer (CRPC). J. Clin. Oncol. 2015, 33, 5000. [Google Scholar] [CrossRef]
- Castration-Resistant Prostate Cancer: Mechanisms, Targets and Treatment. SpringerLink. Available online: https://link.springer.com/chapter/10.1007/978-3-319-99286-0_7 (accessed on 24 May 2023).
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Iannantuono, G.M.; Chandran, E.; Floudas, C.S.; Choo-Wosoba, H.; Butera, G.; Roselli, M.; Gulley, J.L.; Karzai, F. Efficacy and Safety of PARP Inhibitors in Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis of Clinical Trials. Cancer Treat. Rev. 2023, 120, 102623. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The Genomic Landscape of Metastatic Castration-Resistant Prostate Cancers Reveals Multiple Distinct Genotypes with Potential Clinical Impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 175, 889. [Google Scholar] [CrossRef]
- Saraon, P.; Jarvi, K.; Diamandis, E.P. Molecular Alterations during Progression of Prostate Cancer to Androgen Independence. Clin. Chem. 2011, 57, 1366–1375. [Google Scholar] [CrossRef]
- Powell, S.M.; Christiaens, V.; Voulgaraki, D.; Waxman, J.; Claessens, F.; Bevan, C.L. Mechanisms of Androgen Receptor Signalling via Steroid Receptor Coactivator-1 in Prostate. Endocr. Relat. Cancer 2004, 11, 117–130. [Google Scholar] [CrossRef]
- Taplin, M.-E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the Androgen-Receptor Gene in Metastatic Androgen-Independent Prostate Cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Newmark, J.R.; Hardy, D.O.; Tonb, D.C.; Carter, B.S.; Epstein, J.I.; Isaacs, W.B.; Brown, T.R.; Barrack, E.R. Androgen Receptor Gene Mutations in Human Prostate Cancer. Proc. Natl. Acad. Sci. USA 1992, 89, 6319–6323. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W.; Gleave, M.E. Targeting the Adaptive Molecular Landscape of Castration-Resistant Prostate Cancer. EMBO Mol. Med. 2015, 7, 878–894. [Google Scholar] [CrossRef] [PubMed]
- Wilding, G.; Chen, M.; Gelmann, E.P. Aberrant Response in Vitro of Hormone-Responsive Prostate Cancer Cells to Antiandrogens. Prostate 1989, 14, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Taplin, M.-E.; Bubley, G.J.; Ko, Y.-J.; Small, E.J.; Upton, M.; Rajeshkumar, B.; Balk, S.P. Selection for Androgen Receptor Mutations in Prostate Cancers Treated with Androgen Antagonist1. Cancer Res. 1999, 59, 2511–2515. [Google Scholar]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen Receptor Activation in Prostatic Tumor Cell Lines by Insulin-Like Growth Factor-1,Keratinocyte Growth Factor and Epidermal Growth Factor. Eur. Urol. 2017, 27, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Miyazaki, J.; Araki, H.; Yamaoka, M.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel Mutations of Androgen Receptor: A Possible Mechanism of Bicalutamide Withdrawal Syndrome. Cancer Res. 2003, 63, 149–153. [Google Scholar] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging Mechanisms of Resistance to Androgen Receptor Inhibitors in Prostate Cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Tan, J.; Sharief, Y.; Hamil, K.G.; Gregory, C.W.; Zang, D.Y.; Sar, M.; Gumerlock, P.H.; White, R.W.D.; Pretlow, T.G.; Harris, S.E.; et al. Dehydroepiandrosterone Activates Mutant Androgen Receptors Expressed in the Androgen-Dependent Human Prostate Cancer Xenograft CWR22 and LNCaP Cells. Mol. Endocrinol. 1997, 11, 450–459. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A Novel Androgen Receptor Splice Variant Is Up-Regulated during Prostate Cancer Progression and Promotes Androgen Depletion–Resistant Growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef]
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Drabovich, A.P.; Jarvi, K.A.; Diamandis, E.P. Mechanisms of Androgen-Independent Prostate Cancer. EJIFCC 2014, 25, 42–54. [Google Scholar] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen Receptor (AR) Coregulators: An Overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.W.; He, B.; Johnson, R.T.; Ford, O.H.; Mohler, J.L.; French, F.S.; Wilson, E.M. A Mechanism for Androgen Receptor-Mediated Prostate Cancer Recurrence after Androgen Deprivation Therapy. Cancer Res. 2001, 61, 4315–4319. [Google Scholar] [PubMed]
- Zhou, X.E.; Suino-Powell, K.M.; Li, J.; He, Y.; Mackeigan, J.P.; Melcher, K.; Yong, E.-L.; Xu, H.E. Identification of SRC3/AIB1 as a Preferred Coactivator for Hormone-Activated Androgen Receptor. J. Biol. Chem. 2010, 285, 9161–9171. [Google Scholar] [CrossRef] [PubMed]
- Tien, J.C.-Y.; Liu, Z.; Liao, L.; Wang, F.; Xu, Y.; Wu, Y.-L.; Zhou, N.; Ittmann, M.; Xu, J. The Steroid Receptor Coactivator-3 Is Required for the Development of Castration-Resistant Prostate Cancer. Cancer Res. 2013, 73, 3997–4008. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Brzostek, S.; Lee, S.R.; Hollenberg, A.N.; Balk, S.P. Inhibition of the Dihydrotestosterone-Activated Androgen Receptor by Nuclear Receptor Corepressor. Mol. Endocrinol. 2002, 16, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Godoy, A.S.; Sotomayor, P.C.; Villagran, M.; Yacoub, R.; Montecinos, V.P.; McNerney, E.M.; Moser, M.; Foster, B.A.; Onate, S.A. Altered Corepressor SMRT Expression and Recruitment to Target Genes as a Mechanism That Change the Response to Androgens in Prostate Cancer Progression. Biochem. Biophys. Res. Commun. 2012, 423, 564–570. [Google Scholar] [CrossRef]
- Liao, G.; Chen, L.-Y.; Zhang, A.; Godavarthy, A.; Xia, F.; Ghosh, J.C.; Li, H.; Chen, J.D. Regulation of Androgen Receptor Activity by the Nuclear Receptor Corepressor SMRT*. J. Biol. Chem. 2003, 278, 5052–5061. [Google Scholar] [CrossRef]
- Nacusi, L.P.; Tindall, D.J. Targeting 5α-Reductase for Prostate Cancer Prevention and Treatment. Nat. Rev. Urol. 2011, 8, 378–384. [Google Scholar] [CrossRef]
- Dutt, S.S.; Gao, A.C. Molecular Mechanisms of Castration-Resistant Prostate Cancer Progression. Future Oncol. 2009, 5, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.D.; Haugk, K.; Woodke, L.; Nelson, P.; Coleman, I.; Plymate, S.R. Interaction of IGF Signaling and the Androgen Receptor in Prostate Cancer Progression. J. Cell. Biochem. 2006, 99, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, K.; Neuwirt, H.; Cavarretta, I.T.; Bektic, J.; Steiner, H.; Dietrich, H.; Moser, P.L.; Fuchs, D.; Hobisch, A.; Culig, Z. Interleukin-6 Stimulation of Growth of Prostate Cancer in Vitro and in Vivo through Activation of the Androgen Receptor. Endocr. Relat. Cancer 2009, 16, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Reid, A.H.M.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT Pathway for the Treatment of Prostate Cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.H.; Hung, M.C. HER-2/Neu Promotes Androgen-Independent Survival and Growth of Prostate Cancer Cells through the Akt Pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar] [PubMed]
- Graff, J.R.; Konicek, B.W.; McNulty, A.M.; Wang, Z.; Houck, K.; Allen, S.; Paul, J.D.; Hbaiu, A.; Goode, R.G.; Sandusky, G.E.; et al. Increased AKT Activity Contributes to Prostate Cancer Progression by Dramatically Accelerating Prostate Tumor Growth and Diminishing p27Kip1 Expression. J. Biol. Chem. 2000, 275, 24500–24505. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical Implications of PTEN Loss in Prostate Cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Andrieu, C.; Taieb, D.; Baylot, V.; Ettinger, S.; Soubeyran, P.; De-Thonel, A.; Nelson, C.; Garrido, C.; So, A.; Fazli, L.; et al. Heat Shock Protein 27 Confers Resistance to Androgen Ablation and Chemotherapy in Prostate Cancer Cells through eIF4E. Oncogene 2010, 29, 1883–1896. [Google Scholar] [CrossRef]
- Baylot, V.; Katsogiannou, M.; Andrieu, C.; Taieb, D.; Acunzo, J.; Giusiano, S.; Fazli, L.; Gleave, M.; Garrido, C.; Rocchi, P. Targeting TCTP as a New Therapeutic Strategy in Castration-Resistant Prostate Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 2244–2256. [Google Scholar] [CrossRef]
- Cherif, C.; Nguyen, D.T.; Paris, C.; Le, T.K.; Sefiane, T.; Carbuccia, N.; Finetti, P.; Chaffanet, M.; Kaoutari, A.E.; Vernerey, J.; et al. Menin Inhibition Suppresses Castration-Resistant Prostate Cancer and Enhances Chemosensitivity. Oncogene 2022, 41, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Le, T.K.; Cherif, C.; Omabe, K.; Paris, C.; Lannes, F.; Audebert, S.; Baudelet, E.; Hamimed, M.; Barbolosi, D.; Finetti, P.; et al. DDX5 mRNA-Targeting Antisense Oligonucleotide as a New Promising Therapeutic in Combating Castration-Resistant Prostate Cancer. Mol. Ther. 2022, 31, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.; Eder, I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2018, 24, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Cheng, H.; Lin, D.; Liu, L.; Yang, O.; Jia, L.; Fazli, L.; Gleave, M.E.; Wang, Y.; Rennie, P.; et al. The Expression of Glucocorticoid Receptor Is Negatively Regulated by Active Androgen Receptor Signaling in Prostate Tumors. Int. J. Cancer 2015, 136, E27–E38. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.K.; Chugh, N.; Tripathi, M. Neuroendocrine Differentiation of Prostate Cancer—An Intriguing Example of Tumor Evolution at Play. Cancers 2019, 11, 1405. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive Variants of Castration Resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Lin, D.; Wyatt, A.W.; Xue, H.; Wang, Y.; Dong, X.; Haegert, A.; Wu, R.; Brahmbhatt, S.; Mo, F.; Jong, L.; et al. High Fidelity Patient-Derived Xenografts for Accelerating Prostate Cancer Discovery and Drug Development. Cancer Res. 2014, 74, 1272–1283. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 Cooperate to Suppress Prostate Cancer Lineage Plasticity, Metastasis, and Antiandrogen Resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef]
- Deng, X.; Liu, H.; Huang, J.; Cheng, L.; Keller, E.T.; Parsons, S.J.; Hu, C.-D. Ionizing Radiation Induces Prostate Cancer Neuroendocrine Differentiation through Interplay of CREB and ATF2: Implications for Disease Progression. Cancer Res. 2008, 68, 9663–9670. [Google Scholar] [CrossRef] [PubMed]
- Bonkhoff, H. Factors Implicated in Radiation Therapy Failure and Radiosensitization of Prostate Cancer. Prostate Cancer 2012, 2012, 593241. [Google Scholar] [CrossRef] [PubMed]
- Berruti, A.; Dogliotti, L.; Mosca, A.; Bellina, M.; Mari, M.; Torta, M.; Tarabuzzi, R.; Bollito, E.; Fontana, D.; Angeli, A. Circulating Neuroendocrine Markers in Patients with Prostate Carcinoma. Cancer 2000, 88, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Zoubeidi, A.; Selth, L.A. The Epigenetic and Transcriptional Landscape of Neuroendocrine Prostate Cancer. Endocr. Relat. Cancer 2020, 27, R35–R50. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Takeda, D.Y.; Seo, J.-H.; Hwang, J.; Ku, S.Y.; Arafeh, R.; Arnoff, T.; Agarwal, S.; Bell, C.; O’Connor, E.; et al. Reprogramming of the FOXA1 Cistrome in Treatment-Emergent Neuroendocrine Prostate Cancer. Nat. Commun. 2021, 12, 1979. [Google Scholar] [CrossRef]
- Dang, Q.; Li, L.; Xie, H.; He, D.; Chen, J.; Song, W.; Chang, L.S.; Chang, H.-C.; Yeh, S.; Chang, C. Anti-Androgen Enzalutamide Enhances Prostate Cancer Neuroendocrine (NE) Differentiation via Altering the Infiltrated Mast Cells→Androgen Receptor (AR)→miRNA32 Signals. Mol. Oncol. 2015, 9, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal Epigenetic Alterations Drive Metabolic and Neuroendocrine Prostate Cancer Reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. A History of Prostate Cancer Treatment. Nat. Rev. Cancer 2002, 2, 389–396. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Approves Relugolix for Advanced Prostate Cancer; US Food and Drug Administration: Silver Spring, MD, USA, 2021.
- Shore, N.D.; Saad, F.; Cookson, M.S.; George, D.J.; Saltzstein, D.R.; Tutrone, R.; Akaza, H.; Bossi, A.; van Veenhuyzen, D.F.; Selby, B.; et al. Oral Relugolix for Androgen-Deprivation Therapy in Advanced Prostate Cancer. N. Engl. J. Med. 2020, 382, 2187–2196. [Google Scholar] [CrossRef]
- Gamat, M.; McNeel, D.G. Androgen Deprivation and Immunotherapy for the Treatment of Prostate Cancer. Endocr. Relat. Cancer 2017, 24, T297–T310. [Google Scholar] [CrossRef] [PubMed]
- Merseburger, A.S.; Hammerer, P.; Rozet, F.; Roumeguère, T.; Caffo, O.; da Silva, F.C.; Alcaraz, A. Androgen Deprivation Therapy in Castrate-Resistant Prostate Cancer: How Important Is GnRH Agonist Backbone Therapy? World J. Urol. 2015, 33, 1079–1085. [Google Scholar] [CrossRef]
- Heidenreich, A.; Bastian, P.J.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F.; et al. EAU Guidelines on Prostate Cancer. Part II: Treatment of Advanced, Relapsing, and Castration-Resistant Prostate Cancer. Eur. Urol. 2014, 65, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.S.; Roth, B.J.; Dahm, P.; Engstrom, C.; Freedland, S.J.; Hussain, M.; Lin, D.W.; Lowrance, W.T.; Murad, M.H.; Oh, W.K.; et al. Castration-Resistant Prostate Cancer: AUA Guideline. J. Urol. 2013, 190, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.L.; Kantoff, P.W.; Armstrong, A.J.; Bahnson, R.R.; Cohen, M.; D’Amico, A.V.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 1.2014. J. Natl. Compr. Cancer Netw. 2013, 11, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Rove, K.O. Incomplete Testosterone Suppression in Prostate Cancer. N. Engl. J. Med. 2010, 363, 1976. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Page, S.T.; Lin, D.W.; Fazli, L.; Coleman, I.M.; True, L.D.; Knudsen, B.; Hess, D.L.; Nelson, C.C.; Matsumoto, A.M.; et al. Intraprostatic Androgens and Androgen-Regulated Gene Expression Persist after Testosterone Suppression: Therapeutic Implications for Castration-Resistant Prostate Cancer. Cancer Res. 2007, 67, 5033–5041. [Google Scholar] [CrossRef]
- Egan, A.; Dong, Y.; Zhang, H.; Qi, Y.; Balk, S.P.; Sartor, O. Castration-Resistant Prostate Cancer: Adaptive Responses in the Androgen Axis. Cancer Treat. Rev. 2014, 40, 426–433. [Google Scholar] [CrossRef]
- Hager, S.; Ackermann, C.J.; Joerger, M.; Gillessen, S.; Omlin, A. Anti-Tumour Activity of Platinum Compounds in Advanced Prostate Cancer—A Systematic Literature Review. Ann. Oncol. 2016, 27, 975–984. [Google Scholar] [CrossRef]
- Martin, S.K.; Kyprianou, N. Chapter Three—Exploitation of the Androgen Receptor to Overcome Taxane Resistance in Advanced Prostate Cancer. In Advances in Cancer Research; Fisher, P.B., Tew, K.D., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 127, pp. 123–158. [Google Scholar]
- Seruga, B.; Tannock, I.F. Chemotherapy-Based Treatment for Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2011, 29, 3686–3694. [Google Scholar] [CrossRef]
- Pivot, X.; Koralewski, P.; Hidalgo, J.L.; Chan, A.; Gonçalves, A.; Schwartsmann, G.; Assadourian, S.; Lotz, J.P. A Multicenter Phase II Study of XRP6258 Administered as a 1-h i.v. Infusion Every 3 Weeks in Taxane-Resistant Metastatic Breast Cancer Patients. Ann. Oncol. 2008, 19, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; Osoba, D.; Stockler, M.R.; Ernst, D.S.; Neville, A.J.; Moore, M.J.; Armitage, G.R.; Wilson, J.J.; Venner, P.M.; Coppin, C.M.; et al. Chemotherapy with Mitoxantrone plus Prednisone or Prednisone Alone for Symptomatic Hormone-Resistant Prostate Cancer: A Canadian Randomized Trial with Palliative End Points. J. Clin. Oncol. 1996, 14, 1756–1764. Available online: https://ascopubs.org/doi/10.1200/JCO.1996.14.6.1756 (accessed on 25 May 2023). [CrossRef] [PubMed]
- Mitoxantrone. Available online: https://go.drugbank.com/drugs/DB01204 (accessed on 24 May 2023).
- Chemotherapy for Prostate Cancer. Available online: https://www.cancer.org/cancer/types/prostate-cancer/treating/chemotherapy.html (accessed on 15 August 2023).
- Rehman, L.U.; Nisar, M.H.; Fatima, W.; Sarfraz, A.; Azeem, N.; Sarfraz, Z.; Robles-Velasco, K.; Cherrez-Ojeda, I. Immunotherapy for Prostate Cancer: A Current Systematic Review and Patient Centric Perspectives. J. Clin. Med. 2023, 12, 1446. [Google Scholar] [CrossRef] [PubMed]
- Westdorp, H.; Sköld, A.E.; Snijer, B.A.; Franik, S.; Mulder, S.F.; Major, P.P.; Foley, R.; Gerritsen, W.R.; de Vries, I.J.M. Immunotherapy for Prostate Cancer: Lessons from Responses to Tumor-Associated Antigens. Front. Immunol. 2014, 5, 191. [Google Scholar] [CrossRef] [PubMed]
- Dendreon Corporation. Sipuleucel-T. Drugs R D 2006, 7, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Fay, E.K.; Graff, J.N. Immunotherapy in Prostate Cancer. Cancers 2020, 12, 1752. [Google Scholar] [CrossRef] [PubMed]
- Buonerba, C.; Ferro, M.; Di Lorenzo, G. Sipuleucel-T for Prostate Cancer: The Immunotherapy Era Has Commenced. Expert Rev. Anticancer Ther. 2011, 11, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Iranzo, P.; Callejo, A.; Assaf, J.D.; Molina, G.; Lopez, D.E.; Garcia-Illescas, D.; Pardo, N.; Navarro, A.; Martinez-Marti, A.; Cedres, S.; et al. Overview of Checkpoint Inhibitors Mechanism of Action: Role of Immune-Related Adverse Events and Their Treatment on Progression of Underlying Cancer. Front. Med. 2022, 9, 875974. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Shimizu, K.; Sano, T.; Mizuno, K.; Sunada, T.; Makita, N.; Hagimoto, H.; Goto, T.; Sawada, A.; Fujimoto, M.; Ichioka, K.; et al. A Case of Microsatellite Instability-High Clinically Advanced Castration-Resistant Prostate Cancer Showing a Remarkable Response to Pembrolizumab Sustained over at Least 18 Months. Cold Spring Harb. Mol. Case Stud. 2022, 8, a006194. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.N.; Beer, T.M.; Alumkal, J.J.; Slottke, R.E.; Redmond, W.L.; Thomas, G.V.; Thompson, R.F.; Wood, M.A.; Koguchi, Y.; Chen, Y.; et al. A Phase II Single-Arm Study of Pembrolizumab with Enzalutamide in Men with Metastatic Castration-Resistant Prostate Cancer Progressing on Enzalutamide Alone. J. Immunother. Cancer 2020, 8, e000642. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.; Song, L.; Wang, B.Y.; Kalebasty, A.R.; Uchio, E.; Zi, X. Prostate Cancer Immunotherapy: A Review of Recent Advancements with Novel Treatment Methods and Efficacy. Am. J. Clin. Exp. Urol. 2022, 10, 210–233. [Google Scholar] [PubMed]
- Israeli, R.S.; Powell, C.T.; Corr, J.G.; Fair, W.R.; Heston, W.D. Expression of the Prostate-Specific Membrane Antigen. Cancer Res. 1994, 54, 1807–1811. [Google Scholar]
- Palamiuc, L.; Emerling, B.M. PSMA Brings New Flavors to PI3K Signaling: A Role for Glutamate in Prostate Cancer. J. Exp. Med. 2018, 215, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Kaittanis, C.; Andreou, C.; Hieronymus, H.; Mao, N.; Foss, C.A.; Eiber, M.; Weirich, G.; Panchal, P.; Gopalan, A.; Zurita, J.; et al. Prostate-Specific Membrane Antigen Cleavage of Vitamin B9 Stimulates Oncogenic Signaling through Metabotropic Glutamate Receptors. J. Exp. Med. 2017, 215, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-Labeled PSMA I&T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Ng, S.; et al. [177Lu]Lu-PSMA-617 versus Cabazitaxel in Patients with Metastatic Castration-Resistant Prostate Cancer (TheraP): A Randomised, Open-Label, Phase 2 Trial. Lancet 2021, 397, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Agrawal, S.; Gittleman, H.; Fiero, M.H.; Subramaniam, S.; John, C.; Chen, W.; Ricks, T.K.; Niu, G.; Fotenos, A.; et al. FDA Approval Summary: Lutetium Lu 177 Vipivotide Tetraxetan for Patients with Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2023, 29, 1651–1657. [Google Scholar] [CrossRef]
- EMA Pluvicto. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/pluvicto (accessed on 15 May 2023).
- Sartor, A.O.; Morris, M.J.; Messman, R.; Krause, B.J. VISION: An International, Prospective, Open-Label, Multicenter, Randomized Phase III Study of 177Lu-PSMA-617 in the Treatment of Patients with Progressive PSMA-Positive Metastatic Castration-Resistant Prostate Cancer (mCRPC). JCO 2020, 38, TPS259. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Gafita, A.; Marcus, C.; Kostos, L.; Schuster, D.M.; Calais, J.; Hofman, M.S. Predictors and Real-World Use of Prostate-Specific Radioligand Therapy: PSMA and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Baboudjian, M.; Gauthé, M.; Barret, E.; Brureau, L.; Rocchi, P.; Créhange, G.; Dariane, C.; Fiard, G.; Fromont, G.; Beauval, J.-B.; et al. How PET-CT Is Changing the Management of Non-Metastatic Castration-Resistant Prostate Cancer?: Comment La TEP-TDM Peut Modifier La Prise En Charge Du Cancer de La Prostate Non Métastatique Résistant à La Castration? Prog. Urol. 2022, 32, 6S43–6S53. [Google Scholar] [CrossRef] [PubMed]
- Gafita, A.; Rauscher, I.; Weber, M.; Hadaschik, B.; Wang, H.; Armstrong, W.R.; Tauber, R.; Grogan, T.R.; Czernin, J.; Rettig, M.B.; et al. Novel Framework for Treatment Response Evaluation Using PSMA PET/CT in Patients with Metastatic Castration-Resistant Prostate Cancer (RECIP 1.0): An International Multicenter Study. J. Nucl. Med. 2022, 63, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Buteau, J.P.; Martin, A.J.; Emmett, L.; Iravani, A.; Sandhu, S.; Joshua, A.M.; Francis, R.J.; Zhang, A.Y.; Scott, A.M.; Lee, S.-T.; et al. PSMA and FDG-PET as Predictive and Prognostic Biomarkers in Patients given [177Lu]Lu-PSMA-617 versus Cabazitaxel for Metastatic Castration-Resistant Prostate Cancer (TheraP): A Biomarker Analysis from a Randomised, Open-Label, Phase 2 Trial. Lancet Oncol. 2022, 23, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- John, N.; Pathmanandavel, S.; Crumbaker, M.; Counter, W.; Ho, B.; Yam, A.O.; Wilson, P.; Niman, R.; Ayers, M.; Poole, A.; et al. 177Lu-PSMA SPECT Quantitation at 6 Weeks (Dose 2) Predicts Short Progression Free Survival for Patients Undergoing Lu PSMA I&T Therapy. J. Nucl. Med. 2022, 64, 610–615. [Google Scholar] [CrossRef]
- Rosado, M.M.; Pioli, C. Radiotherapy, PARP Inhibition, and Immune-Checkpoint Blockade: A Triad to Overcome the Double-Edged Effects of Each Single Player. Cancers 2023, 15, 1093. [Google Scholar] [CrossRef]
- Khreish, F.; Ebert, N.; Ries, M.; Maus, S.; Rosar, F.; Bohnenberger, H.; Stemler, T.; Saar, M.; Bartholomä, M.; Ezziddin, S. 225Ac-PSMA-617/177Lu-PSMA-617 Tandem Therapy of Metastatic Castration-Resistant Prostate Cancer: Pilot Experience. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 721–728. [Google Scholar] [CrossRef]
- Feuerecker, B.; Tauber, R.; Knorr, K.; Heck, M.; Beheshti, A.; Seidl, C.; Bruchertseifer, F.; Pickhard, A.; Gafita, A.; Kratochwil, C.; et al. Activity and Adverse Events of Actinium-225-PSMA-617 in Advanced Metastatic Castration-Resistant Prostate Cancer After Failure of Lutetium-177-PSMA. Eur. Urol. 2021, 79, 343–350. [Google Scholar] [CrossRef]
- Paschalis, A.; Sheehan, B.; Riisnaes, R.; Rodrigues, D.N.; Gurel, B.; Bertan, C.; Ferreira, A.; Lambros, M.B.K.; Seed, G.; Yuan, W.; et al. Prostate-Specific Membrane Antigen Heterogeneity and DNA Repair Defects in Prostate Cancer. Eur. Urol. 2019, 76, 469–478. [Google Scholar] [CrossRef]
- Bakht, M.K.; Yamada, Y.; Ku, S.-Y.; Venkadakrishnan, V.B.; Korsen, J.A.; Kalidindi, T.M.; Mizuno, K.; Ahn, S.H.; Seo, J.-H.; Garcia, M.M.; et al. Landscape of Prostate-Specific Membrane Antigen Heterogeneity and Regulation in AR-Positive and AR-Negative Metastatic Prostate Cancer. Nat. Cancer 2023, 4, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Foletti, J.-M.; Bardiès, M.; Rocchi, P.; Hicks, R.J.; Haberkorn, U. PSMA-Targeted Radionuclide Therapy and Salivary Gland Toxicity: Why Does It Matter? J. Nucl. Med. 2018, 59, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Devgan, A.; Bansal, I.; Olsavsky, T.D.; Li, S.; Abdelbaki, A.; Kumar, Y. Usefulness of Radium-223 in Patients with Bone Metastases. Proc. Bayl. Univ. Med. Cent. 2017, 30, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Ritter, M.A.; Cleaver, J.E.; Tobias, C.A. High-LET Radiations Induce a Large Proportion of Non-Rejoining DNA Breaks. Nature 1977, 266, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Cislo, P.; Sartor, O.; Vogelzang, N.J.; Coleman, R.E.; O’Sullivan, J.M.; Reuning-Scherer, J.; Shan, M.; Zhan, L.; Parker, C. Patient-Reported Quality-of-Life Analysis of Radium-223 Dichloride from the Phase III ALSYMPCA Study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 868–874. [Google Scholar] [CrossRef]
- Congregado, B.; Rivero, I.; Osmán, I.; Sáez, C.; López, R.M. PARP Inhibitors: A New Horizon for Patients with Prostate Cancer. Biomedicines 2022, 10, 1416. [Google Scholar] [CrossRef]
- Liang, F.; Han, M.; Romanienko, P.J.; Jasin, M. Homology-Directed Repair Is a Major Double-Strand Break Repair Pathway in Mammalian Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 5172–5177. [Google Scholar] [CrossRef]
- FDA Approves Olaparib, Rucaparib to Treat Prostate Cancer—NCI. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2020/fda-olaparib-rucaparib-prostate-cancer (accessed on 12 May 2023).
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Fizazi, K.; Piulats, J.M.; Reaume, M.N.; Ostler, P.; McDermott, R.; Gingerich, J.R.; Pintus, E.; Sridhar, S.S.; Bambury, R.M.; Emmenegger, U.; et al. Rucaparib or Physician’s Choice in Metastatic Prostate Cancer. N. Engl. J. Med. 2023, 388, 719–732. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Approves Olaparib with Abiraterone and Prednisone (or Prednisolone) for BRCA-Mutated Metastatic Castration-Resistant Prostate Cancer; US Food and Drug Administration: Silver Spring, MD, USA, 2023.
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in Patients with Metastatic Castration-Resistant Prostate Cancer and DNA Repair Gene Defects (GALAHAD): A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- De Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib Monotherapy in Metastatic Castration-Resistant Prostate Cancer with DNA Repair Alterations (TALAPRO-1): An Open-Label, Phase 2 Trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.A.; Carles, J.; Fay, A.P.; Matsubara, N.; Heinrich, D.; Szczylik, C.; Giorgi, U.D.; Joung, J.Y.; Fong, P.C.C.; et al. Talazoparib plus Enzalutamide in Men with First-Line Metastatic Castration-Resistant Prostate Cancer (TALAPRO-2): A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 402, 291–303. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Approves Niraparib and Abiraterone Acetate plus Prednisone for BRCA-Mutated Metastatic Castration-Resistant Prostate Cancer; US Food and Drug Administration: Silver Spring, MD, USA, 2023.
- Slootbeek, P.H.J.; Overbeek, J.K.; Ligtenberg, M.J.L.; van Erp, N.P.; Mehra, N. PARPing up the Right Tree; an Overview of PARP Inhibitors for Metastatic Castration-Resistant Prostate Cancer. Cancer Lett. 2023, 216367. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.K.; Duong, Q.H.; Baylot, V.; Fargette, C.; Baboudjian, M.; Colleaux, L.; Taïeb, D.; Rocchi, P. Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments. Cancers 2023, 15, 5047. https://doi.org/10.3390/cancers15205047
Le TK, Duong QH, Baylot V, Fargette C, Baboudjian M, Colleaux L, Taïeb D, Rocchi P. Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments. Cancers. 2023; 15(20):5047. https://doi.org/10.3390/cancers15205047
Chicago/Turabian StyleLe, Thi Khanh, Quang Hieu Duong, Virginie Baylot, Christelle Fargette, Michael Baboudjian, Laurence Colleaux, David Taïeb, and Palma Rocchi. 2023. "Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments" Cancers 15, no. 20: 5047. https://doi.org/10.3390/cancers15205047