
Modulation of JAK-STAT Signaling by LNK: A Forgotten Oncogenic Pathway in Hormone Receptor-Positive Breast Cancer


Abstract
:1. Introduction
2. Hormone Receptor-Positive Breast Cancer
3. JAK-STAT Signaling Pathway
3.1. The JAK Family
3.2. The STAT Family
4. JAK-STAT Signaling in Hormone Receptor-Positive Breast Cancer
4.1. Activation of the JAK-STAT Pathway by IL-6
4.2. Activation of the JAK-STAT Pathway by PRL
5. Adaptor Molecule LNK as an Inhibitor of JAK-STAT
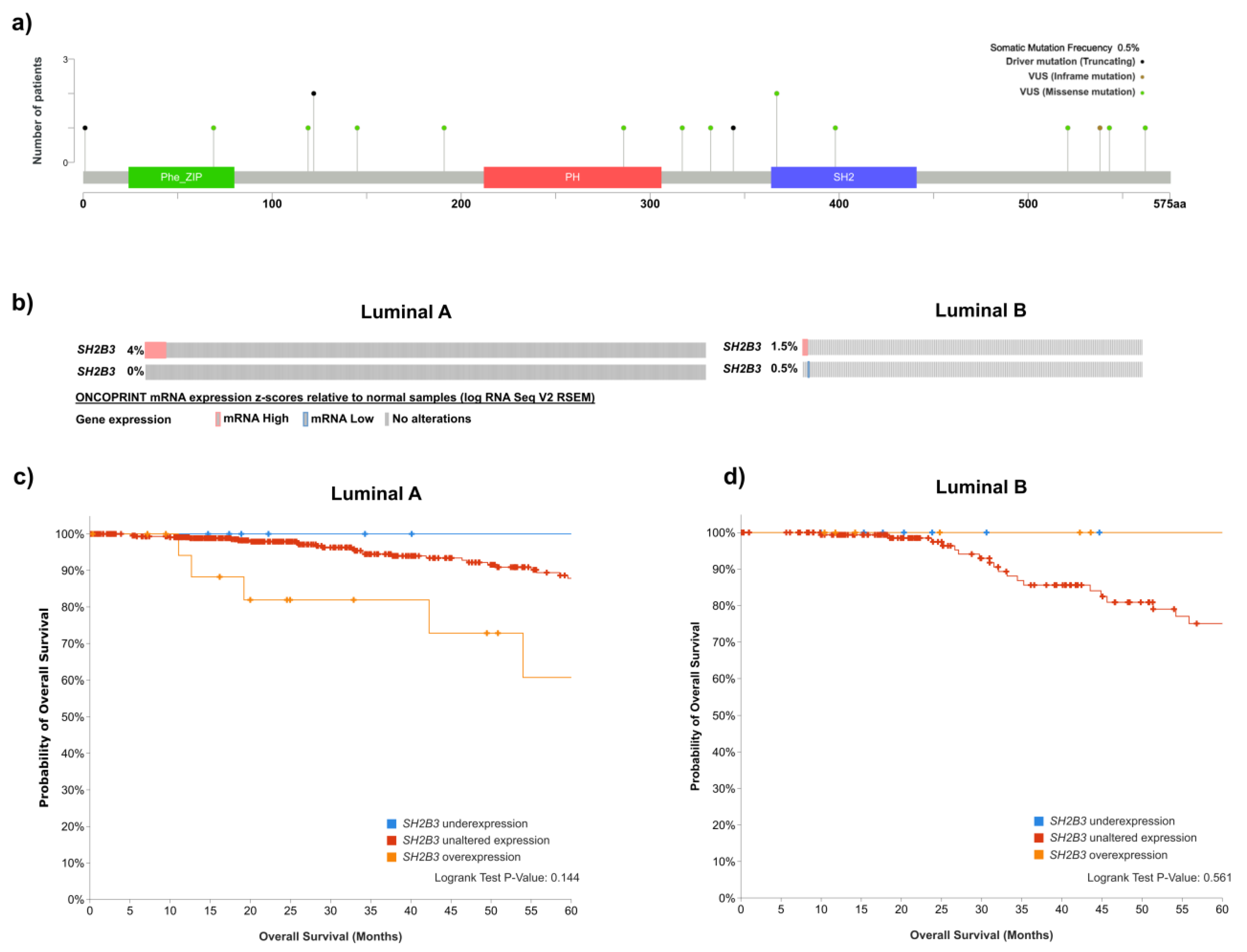
6. LNK in Hormone Receptor-Positive Breast Cancer
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Mallepell, S.; Krust, A.; Chambon, P.; Brisken, C. Paracrine Signaling through the Epithelial Estrogen Receptor Alpha Is Required for Proliferation and Morphogenesis in the Mammary Gland. Proc. Natl. Acad. Sci. USA 2006, 103, 2196–2201. [Google Scholar] [CrossRef] [PubMed]
- Brisken, C.; Park, S.; Vass, T.; Lydon, J.P.; O’Malley, B.W.; Weinberg, R.A. A Paracrine Role for the Epithelial Progesterone Receptor in Mammary Gland Development. Proc. Natl. Acad. Sci. USA 1998, 95, 5076–5081. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The Nuclear Receptor Superfamily: The Second Decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Haider, K.; Neha, K.; Yar, M.S. Aromatase Inhibitors: Role in Postmenopausal Breast Cancer. Arch. Pharm. 2020, 353, 2000081. [Google Scholar] [CrossRef]
- AlFakeeh, A.; Brezden-Masley, C. Overcoming Endocrine Resistance in Hormone Receptor-Positive Breast Cancer. Curr. Oncol. 2018, 25 (Suppl. S1), S18–S27. [Google Scholar] [CrossRef]
- Stevens, L.E.; Peluffo, G.; Qiu, X.; Temko, D.; Fassl, A.; Li, Z.; Trinh, A.; Seehawer, M.; Jovanovic, B.; Aleckovic, M.; et al. JAK-STAT Signaling in Inflammatory Breast Cancer Enables Chemotherapy-Resistant Cell States. Cancer Res. 2023, 83, 264–284. [Google Scholar] [CrossRef]
- Shao, F.; Pang, X.; Baeg, G.H. Targeting the JAK/STAT Signaling Pathway for Breast Cancer. Curr. Med. Chem. 2021, 28, 5137–5151. [Google Scholar] [CrossRef]
- Morris, R.; Butler, L.; Perkins, A.; Kershaw, N.J.; Babon, J.J. The Role of LNK (SH2B3) in the Regulation of JAK-STAT Signalling in Haematopoiesis. Pharmaceuticals 2021, 15, 24. [Google Scholar] [CrossRef]
- Trapani, D.; Ginsburg, O.; Fadelu, T.; Lin, N.U.; Hassett, M.; Ilbawi, A.M.; Anderson, B.O.; Curigliano, G. Global Challenges and Policy Solutions in Breast Cancer Control. Cancer Treat. Rev. 2022, 104, 102339. [Google Scholar] [CrossRef]
- Knight, S.R.; Chu, K.; Lapitan, M.C.; Dare, A.J.; Pius, R.; Shaw, C.A.; Drake, T.M.; Norman, L.; Ademuyiwa, A.O.; Adisa, A.O.; et al. Effects of Hospital Facilities on Patient Outcomes after Cancer Surgery: An International, Prospective, Observational Study. Lancet Glob. Health 2022, 10, e1003–e1011. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. GLOBOCAN. Cancer Tomorrow. Available online: https://gco.iarc.fr/tomorrow/en (accessed on 27 June 2023).
- Maringe, C.; Spicer, J.; Morris, M.; Purushotham, A.; Nolte, E.; Sullivan, R.; Rachet, B.; Aggarwal, A. The Impact of the COVID-19 Pandemic on Cancer Deaths Due to Dalys in Diagnosis in England, UK: A National, Population-Based, Modelling Study. Lancet Oncol. 2020, 21, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.H.; Ellis, I.; Allison, K.; Brogi, E.; Fox, S.B.; Lakhani, S.; Lazar, A.J.; Morris, E.A.; Sahin, A.; Salgado, R.; et al. The 2019 World Health Organization Classification of Tumours of the Breast. Histopathology 2020, 77, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; Ellis, I.O.; Schnitt, S.J.; Tan, P.H.; van de Vijver, M.J. WHO Classification of Tumours of the Breast, 4th ed.; IARC Press: Lyon, France, 2012. [Google Scholar]
- Singhai, R.; Patil, V.W.; Patil, A.V. Immunohistochemical (IHC) HER2/neu and Fluorescent in Situ Hybridization (FISH) Gene Amplification of Breast Cancer in Indian Women. Asian Pac. J. Cancer Prev. 2011, 12, 179–183. [Google Scholar] [PubMed]
- Wesola, M.; Jelén, M. A Comparison of IHC and FISH Cytogenetic Methods in the Evaluation of HER2 Status in Breast Cancer. Adv. Clin. Exp. Med. 2015, 24, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Muñoz, A.; Garcia-Tapiador, A.M.; Martinez-Ortega, E.; Dueñas-Garcia, R.; Jaen-Morago, A.; Ortega-Granados, A.L.; Fernandez-Navarro, M.; de la Torre-Cabrera, C.; Dueñas, B.; Rueda, A.I.; et al. Tumour Molecular Subtyping According to Hormone Receptors and HER2 Status Defines Different Pathological Complete Response to Neoadjuvant Chemotherapy in Patients with Locally Advanced Breast Cancer. Clin. Transl. Oncol. 2008, 10, 646–653. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Weigelt, B.; Mackay, A.; A’hern, R.; Natrajan, R.; Tan, D.S.P.; Dowsett, M.; Ashworth, A.; Reis-Filho, J.S. Breast Cancer Molecular Profiling with Single Sample Predictors: A Retrospective Analysis. Lancet Oncol. 2010, 11, 339–349. [Google Scholar] [CrossRef]
- Mackay, A.; Weigelt, B.; Grigoriadis, A.; Kreike, B.; Natrajan, R.; A’hern, R.; Tan, D.S.P.; Dowsett, M.; Ashworth, A.; Reis-Filho, J.S. Microarray-Based Class Discovery for Molecular Classification of Breast Cancer: Analysis of Interobserver Agreement. J. Natl. Cancer Inst. 2011, 103, 662–673. [Google Scholar] [CrossRef]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef]
- Andreopoulou, E.; Schweber, S.J.; Sparano, J.A.; McDaid, H.M. Therapies for Triple Negative Breast Cancer. Expert. Opin. Pharmacother. 2015, 16, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Pienkowski, T.; Im, Y.H.; Roman, L.; Tseng, L.M.; Liu, M.C.; Lluch, A.; Staroslawska, E.; de la Haba-Rodriguez, J.; Im, S.A.; et al. Efficacy and Safety of Neoadyuvant Pertuzumab and Trastuzumab in Women with Locally Advanced, Inflammatory, or Early HER2-Positive Breast Cancer (NeoSphere): A Randomised Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2012, 13, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptor in Breast Cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J. Personalizing the Treatment of Women with Early Breast Cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; Anderson, G.L.; Aragaki, A.K.; Manson, J.A.E.; Stefanick, M.L.; Pan, K.; Barrington, W.; Kuller, L.H.; Simon, M.S.; Lane, D.; et al. Association of Menopausal Hormone Therapy with Breast Cancer Incidence and Mortality during Long-Term Follow-Up of the Women’s Health Initiative Randomized Clinical Trials. JAMA 2020, 324, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Pfeiffer, R.M.; Gierach, G.L.; Falk, R.T. Use of Postmenopausal Hormone Therapies and Risk of Histology- and Hormone Receptor-Defined Breast Cancer: Results from a 15-Year Prospective Analysis of NIH-AARP Cohort. Breast Cancer Res. 2020, 22, 129. [Google Scholar] [CrossRef]
- Pupo, M.; Maggiolini, M.; Musti, A.M. GPER Mediates Non-Genomic Effects of Estrogen. Methods Mol. Biol. 2016, 1366, 471–488. [Google Scholar]
- Walter, P.; Green, S.; Greene, G.; Krust, A.; Bornert, J.M.; Jeltsch, J.M.; Staub, A.; Jensen, E.; Scrace, G.; Waterfield, M.; et al. Cloning of the Human Estrogen Receptor cDNA. Proc. Natl. Acad. Sci. USA 1985, 82, 7889–7893. [Google Scholar] [CrossRef]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.-A. Differential Expression of Estrogen Receptor α, β1, and β2 in Lobular and Ductal Breast Cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef]
- Stender, J.D.; Frasor, J.; Komm, B.; Chang, K.C.N.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen-Regulated Gene Networks in Human Breast Cancer Cells: Involvement of E2F1 in the Regulation of Cell Proliferation. Mol. Endocrinol. 2007, 21, 2112–2123. [Google Scholar] [CrossRef]
- Bourdeau, V.; Deschênes, J.; Laperrière, D.; Aid, M.; White, J.; Mader, S. Mechanisms of Primary and Secondary Estrogen Target Gene Regulation in Breast Cancer Cells. Nucleic Acids Res. 2007, 36, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Millour, J.; Constantinidou, D.; Stavropoulou, A.V.; Wilson, M.; Myatt, S.S.; Kwok, J.M.-M.; Sivanandan, K.; Coombes, R.C.; Medema, R.; Hartman, J.; et al. FOXM1 Is a Transcriptional Target of ERα and Has a Critical Role in Breast Cancer Endocrine Sensitivity and Resistance. Oncogene 2010, 29, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- JavanMoghadam, S.; Weihua, Z.; Hunt, K.K.; Keyomarsi, K. Estrogen Receptor Alpha Is Cell Cycle-Regulated and Regulates the Cell Cycle in a Ligand-Dependent Fashion. Cell Cycle 2016, 15, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Speirs, V.; Malone, C.; Walton, D.S.; Kerin, M.J.; Atkin, S.L. Increased Expression of Estrogen Receptor β mRNA in Tamoxifen-Resistant Breast Cancer Patients. Cancer Res. 1999, 59, 5421–5424. [Google Scholar]
- Markey, G.C.; Cullen, R.; Diggin, P.; Hill, A.D.K.; Mc Dermott, E.W.; O’Higgins, N.J.; Duffy, M.J. Estrogen Receptor-β mRNA Is Associated with Adverse Outcome in Patients with Breast Cancer. Tumor Biol. 2009, 30, 171–175. [Google Scholar] [CrossRef]
- Shaaban, A.M.; Green, A.R.; Karthik, S.; Alizadeh, Y.; Hughes, T.A.; Harkins, L.; Ellis, I.O.; Robertson, J.F.; Paish, E.C.; Saunders, P.T.K.; et al. Nuclear and Cytoplasmic Expression of ERbeta1, ERbeta2, and ERbeta5 Identifies Distinct Prognostic Outcome for Breast Cancer Patients. Clin. Cancer Res. 2008, 14, 5228–5235. [Google Scholar] [CrossRef]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Estrogen Receptor Alpha and Beta in Health and Disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef]
- Chen, P.; Li, B.; Ou-Yang, L. Role of Estrogen Receptors in Health and Disease. Front. Endocrinol. 2022, 13, 839005. [Google Scholar] [CrossRef]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty Years of the G Protein-Coupled Estrogen Receptor GPER: Historical and Personal Perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. Estrogen Biology: New Insights into GPER Function and Clinical Opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef]
- Treeck, O.; Schüler-Toprak, S.; Ortmann, O. Estrogen Actions in Triple-Negative Breast Cancer. Cells 2020, 9, 2358. [Google Scholar] [CrossRef] [PubMed]
- Samartzis, E.P.; Noske, A.; Meisel, A.; Varga, Z.; Fink, D.; Imesch, P. The G Protein-Coupled Estrogen Receptor (GPER) Is Expressed in Two Different Subcellular Localizations Reflecting Distinct Tumor Properties in Breast Cancer. PLoS ONE 2014, 9, e83296. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Cupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Quinn, J.A.; Graeber, C.T.; Frackelton, A.R., Jr.; Kim, M.; Schwarzbauer, J.E.; Filardo, E.J. Coordinate Regulation of Estrogen-Mediated Fibronectin Matrix Assembly and Epidermal Growth Factor Receptor Transactivation by the G Protein-Coupled Receptor, GPR30. Mol. Endocrinol. 2009, 23, 1052–1064. [Google Scholar] [CrossRef]
- Yu, T.; Liu, M.; Luo, H.; Wu, C.; Tang, X.; Tang, S.; Hu, P.; Yan, Y.; Wang, Z.; Tu, G. GPER Mediates Enhanced Cell Viability and Motility Via Non-Genomic Signaling Induced by 17β-Estradiol in Triple-Negative Breast Cancer (TNBC) Cells. J. Steroid Biochem. Mol. Biol. 2014, 143, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, T.; Weibenborn, C.; Poehlmann, A.; Lemke, A.; Semczuk, A.; Roessner, A.; Costa, S.D.; Kalinski, T.; Ignatov, A. GPER-1 Expression Decreases during Breast Cancer Tumorigenesis. Cancer Investig. 2013, 31, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, M.; Hartman, L.; Grabau, D.; Fornander, T.; Malmström, P.; Nordenskjöld, B.; Sgroi, D.C.; Skoog, L.; Stål, O.; Leeb-Lundberg, L.M.F.; et al. Lack of G Protein-Coupled Estrogen Receptor (GPER) in the Plasma Membrane Is Associated with Excellent Long-Term Prognosis in Breast Cancer. Breast Cancer Res. Treat. 2014, 145, 61–71. [Google Scholar] [CrossRef]
- Mo, Z.; Liu, M.; Yang, F.; Luo, H.; Li, Z.; Tu, G.; Yang, G. GPR30 as an Initiator of Tamoxifen Resistance in Hormone-Dependent Breast Cancer. Breast Cancer Res. 2013, 15, R114. [Google Scholar] [CrossRef]
- Liang, S.; Chen, Z.; Jiang, G.; Zhou, Y.; Liu, Q.; Su, Q.; Wei, W.; Du, J.; Wang, H. Activation of GPER Suppresses Migration and Angiogenesis of Triple Negative Breast Cancer via Inhibition of NF-κB/IL-6 Signals. Cancer Lett. 2017, 386, 12–23. [Google Scholar] [CrossRef]
- Okamoto, M.; Mizukami, Y. GPER Negatively Regulates TNFα-Induced IL-6 Production in Human Breast Cancer Cells via NF-κB Pathway. Endocr. J. 2016, 63, 485–493. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Aittomäki, S.; Pesu, M. Therapeutic Targeting of the Jak/STAT Pathway. Basic Clin. Pharm. Toxicol. 2014, 114, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edge Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Wilks, A.F.; Harpur, A.G.; Kurban, R.R.; Ralph, S.J.; Zücher, G.; Ziemiecki, A. Two Novel Protein-Tyrosine Kinases, each with a Second Phosphotransferase-Related Catalytic Domain, Define a New Class of Protein Kinase. Mol. Cell. Biol. 1991, 11, 2057–2065. [Google Scholar]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK-Receptor Interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Frank, S.J.; Yi, W.; Zhao, Y.; Goldsmith, J.F.; Gilliland, G.; Jiang, J.; Sakai, I.; Kraft, A.S. Regions of the JAK2 Tyrosine Kinase Required for Coupling to the Growth Hormone Receptor. Biol. Chem. 1995, 270, 14776–14785. [Google Scholar] [CrossRef]
- Velazquez, L.; Mogensen, K.E.; Barbieri, G.; Fellous, M.; Uzé, G.; Pellegrini, S. Distinct Domains of the Protein Tyrosine Kinase Tyk2 Required for Binding of Interferon-Alpha/Beta and for Signal Transduction. J. Biol. Chem. 1995, 270, 3327–3334. [Google Scholar] [CrossRef]
- Rodig, S.J.; Meraz, M.A.; White, J.M.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef]
- Schindler, C.; Strehlow, I. Cytokines and STAT Signaling. Adv. Pharm. 2000, 47, 113–174. [Google Scholar]
- Russell, S.M.; Johnston, J.A.; Noguchi, M.; Kawamura, M.; Bacon, C.M.; Friedmann, M.; Berg, M.; McVicar, D.W.; Witthuhn, B.A.; Silvennoinen, O.; et al. Interaction of IL-2R Beta and Gamma c Chains with Jak1 and Jak3: Implications for XSCID and XCID. Science 1994, 266, 1042–1045. [Google Scholar] [CrossRef]
- Seto, Y.; Nakajima, H.; Suto, A.; Shimoda, K.; Saito, Y.; Nakayama, K.I.; Iwamoto, I. Enhanced Th2 Cell-Mediated Allergic Inflammation in Tyk2-Deficient Mice. J. Immunol. 2003, 170, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Watford, W.T.; O’Sheas, J.J. Human Tyk2 Kinase Deficiency: Another Primary Immunodeficiency Syndrome. Immunity 2006, 25, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Ehret, G.B.; Reichenbach, P.; Schindler, U.; Horvath, C.M.; Fritz, S.; Nabholz, M.; Bucher, P. DNA Binding Specificity of Different STAT Proteins. Comparison of In Vitro Specificity with Natural Target Sites. J. Biol. Chem. 2001, 276, 6675–6688. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, A.; Karlberg, I.; Nilsson, K.; Öberg, F. Ser727/Tyr701-Phosphorylated Stat1 Is Required for the Regulation of c-Myc, Cyclins, and p27Kip1 Associated with ATRA-Induced G0/G1 Arrest of U-937 Cells. Blood 2003, 102, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, A.; Nilsson, K.; Öberg, F. Phosphorylation-Deficient Stat1 Inhibits Retinoic Acid-Induced Differentiation and Cell Cycle Arrest in U-937 Monoblasts. Blood 2000, 96, 2870–2878. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Fu, X.Y.; Plate, J.; Chong, A.S. IFN-Gamma Induces Cell Growth Inhibition by Fas-Mediated Apoptosis: Requirement of STAT1 Protein for Up-Regulation of Fas and FasL Expression. Cancer Res. 1998, 58, 2832–2837. [Google Scholar]
- Stephanou, A.; Brar, B.K.; Knight, R.A.; Latchman, D.S. Opposing Actions of STAT-1 and STAT-3 on the Bcl-2 and Bcl-x Promoters. Cell Death Differ. 2000, 7, 329–330. [Google Scholar] [CrossRef]
- Najjar, I.; Deglesne, P.A.; Schischmanoff, P.O.; Fabre, E.E.; Boisson-Dupuis, S.; Nimmerjahn, F.; Bornkamm, G.W.; Dusanter-Fourt, I.; Fagard, R.J. STAT1-Dependent IgG Cell-Surface Expression in a Human B Cell Line Derived from a STAT1-Deficient Patient. Leukoc. Biol. 2010, 87, 1145–1152. [Google Scholar] [CrossRef]
- Wang, Y.; Song, Q.; Huang, W.; Lin, Y.; Wang, X.; Wang, C.; Willard, B.; Zhao, C.; Nan, J.; Holvey-Bates, E.; et al. A Virus-Induced Conformational Switch of STAT1-STAT2 Dimers Boosts Antiviral Defenses. Cell Res. 2021, 31, 206–218. [Google Scholar] [CrossRef]
- Park, C.; Li, S.; Cha, E.; Schindler, C. Immune Response in Stat2 Knockout Mice. Immunity 2000, 13, 795–804. [Google Scholar] [CrossRef]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 Signaling in Immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Thieu, V.T.; Yu, Q.; Chang, H.-C.; Yeh, N.; Nguyen, E.T.; Sehra, S.; Kaplan, M.H. Signal Transducer and Activator of Transcription 4 Is Required for the Transcription Factor T-bet to Promote T Helper 1 Cell-Fate Determination. Immunity 2008, 29, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Mott, K.R.; Ghiasi, H. Involvement of STAT4 in IgG Subtype Switching and Ocular HSV-1 Replication in Mice. Mol. Vis. 2010, 16, 98–104. [Google Scholar] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Lin, J.X.; Du, N.; Li, P.; Kazemian, M.; Gebregiorgis, T.; Spolski, R.; Leonard, W.J. Critical Functions for STAT5 Tetramers in the Maturation and Survival of Natural Killer Cells. Nat. Commun. 2017, 8, 1320. [Google Scholar] [CrossRef]
- Soldaini, E.; John, S.; Moro, S.; Bollenbacher, J.; Schindler, U.; Leonard, W.J. DNA Binding Site Selection of Dimeric and Tetrameric Stat5 Proteins Reveals a Large Repertoire of Divergent Tetrameric Stat5a Binding Sites. Mol. Cell. Biol. 2000, 20, 389–401. [Google Scholar] [CrossRef]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5 Is Mandatory for Adult Mammary Gland Development and Lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 Is Required for Mediating Responses to IL-4 and for Development of Th2 Cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef]
- Huang, C.Z.; Zou, D.; Yang, J.; Qiao, H.L. Polymorphism of STAT6 and Specific Serum IgE Levels in Patients with Penicillin Allergy. Int. J. Clin. Pharmacol. Ther. 2012, 50, 461–467. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive Activation of STAT3 in Breast Cancer Cells: A Review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Bièche, I.; Lerebours, F.; Tozlu, S.; Espie, M.; Marty, M.; Lidereau, R. Molecular Profiling of Inflammatory Breast Cancer: Identification of a Poor-Prognosis Gene Expression Signature. Clin. Cancer Res. 2004, 10, 6789–6795. [Google Scholar] [CrossRef] [PubMed]
- Speirs, V.; Kerin, M.J.; Walton, D.S.; Newton, C.J.; Desai, S.B.; Atkin, S.L. Direct Activation of Oestrogen Receptor-Alpha by Interleukin-6 in Primary Cultures of Breast Cancer Epithelial Cells. Br. J. Cancer 2000, 82, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Matsuda, T.; Junicho, A.; Kishi, H.; Saatcioglu, F.; Muraguchi, A. Cross-Talk, between Signal Transducer and Activator of Transcription 3 and Estrogen Receptor Signaling. FEBS Lett. 2000, 486, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Siersbæk, R.; Scabia, V.; Nagarajan, S.; Chernukhin, I.; Papachristou, E.K.; Broome, R.; Johnston, S.J.; Joosten, S.E.P.; Green, A.R.; Kumar, S.; et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor α Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020, 38, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, J.; Lee, Y.-H.; Eom, M.; Choi, J. Interleukin-6/STAT3 Signalling Regulates Adipocyte Induced Epithelial-Mesenchimal Transition in Breast Cancer Cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- Yang, L.; Han, S.; Sun, Y. An IL6-STAT3 Loop Mediates Resistance to PI3K Inhibitors by Inducing Epithelial-Mesenchymal Transition and Cancer Stem Cell Expansion in Human Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2014, 453, 582–587. [Google Scholar] [CrossRef]
- Kettner, N.M.; Vijayaraghavan, S.; Durak, M.G.; Bui, T.; Kohansal, M.; Ha, M.J.; Liu, B.; Rao, X.; Wang, J.; Yi, M.; et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin. Cancer Res. 2019, 25, 3996–4013. [Google Scholar] [CrossRef]
- Xing, J.; Li, J.; Fu, L.; Gai, J.; Guan, J.; Li, Q. SIRT4 Enhances the Sensitivity of ER-Positive Breast Cancer to Tamoxifen by Inhibiting the IL-6/STAT3 Signal Pathway. Cancer Med. 2019, 8, 7086–7097. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Tsoi, H.; Man, E.P.S.; Chau, K.M.; Khoo, U.-S. Targeting the IL-6/STAT3 Signalling Cascade to Reverse Tamoxifen Resistance in Estrogen Receptor Positive Breast Cancer. Cancers 2021, 13, 1511. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, M.; Sun, Z.; Ye, Y.; Han, X.; Qin, Y.; Liu, S. Wenshen Zhuanggu Formula Mitigates Breast Cancer Bone Metastasis through the Signaling Crosstalk among the Jagged1/Notch, TGF-β and IL-6 Signaling Pathways. J. Ethnopharmacol. 2019, 232, 145–154. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, X.; Feng, Y.; Zheng, L.; Jian, J. Selenium Donors Inhibits Osteoclastogenesis through Inhibiting IL-6 and Plays a Pivotal Role in Bone Metastasis from Breast Cancer. Cancer Toxicol. Res. 2020, 9, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Clevenger, C.V.; Chang, W.P.; Ngo, W.; Pasha, T.L.; Montone, K.T.; Tomaszewski, J.E. Expression of Prolactin and Prolactin Receptor in Human Breast Carcinoma. Evidence for an Autocrine/Paracrine Loop. Am. J. Pathol. 1995, 146, 695–705. [Google Scholar] [PubMed]
- Reynolds, C.; Montone, K.T.; Powell, C.M.; Tomaszewski, J.E.; Clevenger, C.V. Expression of Prolactin and Its Receptor in Human Breast Carcinoma. Endocrinology 1997, 138, 5555–5560. [Google Scholar] [CrossRef]
- Tworoger, S.S.; Eliassen, A.H.; Zhang, X.; Qian, J.; Sluss, P.M.; Rosner, B.A.; Hankinson, S.E. A 20-Year Prospective Study of Plasma Prolactin as a Risk Marker of Breast Cancer Development. Cancer Res. 2013, 73, 4810–4819. [Google Scholar] [CrossRef]
- Tikk, K.; Sookthai, D.; Johnson, T.; Rinaldi, S.; Romieu, I.; Tjonneland, A.; Olsen, A.; Overvad, K.; Clavel-Chapelon, F.; Baglietto, L.; et al. Circulating Prolactin and Breast Cancer Risk among Pre- and Postmenopausal Women in the EPIC Cohort. Ann. Oncol. 2014, 25, 1422–1428. [Google Scholar] [CrossRef]
- Sutherland, A.; Forsyth, A.; Cong, Y.; Grant, L.; Juan, T.-H.; Lee, J.K.; Klimowicz, A.; Petrillo, S.K.; Hu, J.; Chan, A.; et al. The Role of Prolactin in Bone Metastasis and Breast Cancer Cell-Mediated Osteoclast Differentiation. J. Natl. Cancer Inst. 2015, 108, dvj338. [Google Scholar] [CrossRef]
- Shemanko, C.S. Prolactin Receptor in Breast Cancer: Marker for Metastatic Risk. J. Mol. Endocrinol. 2016, 57, R153–R165. [Google Scholar] [CrossRef]
- Sakamoto, K.; Triplett, A.A.; Schuler, L.A.; Wagner, K.U. Janus Kinase 2 Is Required for the Initiation but Not Maintenance of Prolactin-Induced Mammary Cancer. Oncogene 2010, 29, 5359–5369. [Google Scholar] [CrossRef]
- Chen, K.H.; Walker, A.M. Prolactin Inhibits a Major Tumor-Suppressive Function of Wild Type BRCA1. Cancer Lett. 2016, 375, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Peck, A.R.; Witkiewicz, A.K.; Liu, C.; Stringer, G.A.; Klimowicz, A.C.; Pequignot, E.; Freydin, B.; Tran, T.H.; Yang, N.; Rosenberg, A.L.; et al. Loss of Nuclear Localized and Tyrosine Phosphorylated Stat5 in Breast Cancer Predicts Poor Clinical Outcome and Increased Risk of Antiestrogen Therapy Failure. J. Clin. Oncol. 2011, 29, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, F.; Radu, T.B.; Orlova, A.; Qadree, A.K.; de Araujo, E.D.; Israelina, J.; Valent, P.; Mustjoki, S.M.; Herling, M.; Moriggl, R.; et al. JAK-STAT Core Cancer Pathway: An Integrative Cancer Interactome Analysis. J. Cell. Mol. Med. 2022, 26, 2049–2062. [Google Scholar] [CrossRef]
- Trengove, M.C.; Ward, A.C. SOCS proteins in development and disease. Am. J. Clin. Exp. Immunol. 2013, 2, 1–29. [Google Scholar]
- Shuai, K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006, 16, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatase in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Huang, X.; Li, Y.; Tanaka, K.; Moore, K.G.; Hayashi, J.I. Cloning and Characterization of Lnk, a Signal Transduction Protein that Links T-cell Receptor Activation Signal to Phospholipase C Gamma 1, Grb2, and Phosphatidylinositol 3-Kinase. Proc. Natl. Acad. Sci. USA 1995, 92, 11618–11622. [Google Scholar] [CrossRef]
- Li, Y.; He, X.; Schembri-King, J.; Jakes, S.; Hayashi, J. Cloning and Characterization of Human Lnk, an Adaptor Protein with Pleckstrin Homology and Src Homology 2 Domains that Can Inhibit T Cell Activation. J. Immunol. 2000, 164, 5199–5206. [Google Scholar] [CrossRef]
- Harlan, J.E.; Hajduk, P.J.; Yoon, H.S.; Fesik, S.W. Pleckstrin Homology Domains Bind to Phosphatidylinositol-4,5-Bisphosphate. Nature 1994, 371, 168–170. [Google Scholar] [CrossRef]
- Takaki, S.; Morita, H.; Tezuka, Y.; Takatsu, K. Enhanced Hematopoiesis by Hematopoietic Progenitor Cells Lacking Intracellular Adaptor Protein, Lnk. J. Exp. Med. 2002, 195, 151–160. [Google Scholar] [CrossRef]
- Takaki, S.; Sauer, K.; Iritani, B.M.; Chien, S.; Ebihara, Y.; Tsuji, K.; Takatsu, K.; Perlmutter, R.M. Control of B Cell Production by the Adaptor Protein Lnk: Definition of a Conserved Family of Signal-Modulating Proteins. Immunity 2000, 13, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Takaki, S.; Tezuka, Y.; Sauer, K.; Kubo, C.; Kwon, S.M.; Armstead, E.; Nakao, K.; Katsuki, M.; Perlmutter, R.M.; Takatsu, K. Impaired Lymphopoiesis and Altered B Cell Subpopulations in Mice Overexpressing Lnk Adaptor Protein. J. Immunol. 2003, 170, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Lodish, H.F. Lnk Inhibits Tpo-Mpl Signaling and Tpo-Mediated Megakaryocytopoiesis. J. Exp. Med. 2004, 200, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Zhang, Y.; Ellyard, J.I.; Vinuesa, C.G.; Murphy, J.M.; Laktyushin, A.; Kershaw, N.J.; Babon, J.J. Structural and Functional Analysis of Target Recognition by the Lymphocyte Adaptor Protein LNK. Nat. Commun. 2021, 12, 6110. [Google Scholar] [CrossRef]
- Jiang, J.; Balcerek, J.; Rozenova, K.; Cheng, Y.; Bersenev, A.; Wu, C.; Song, Y.; Tong, W. 14-3-3 Regulates the LNK/JAK2 Pathway in Mouse Hematopoietic Stem and Progenitor Cells. J. Clin. Investig. 2012, 122, 2079–2091. [Google Scholar] [CrossRef]
- Devallière, J.; Chatelais, M.; Fitau, J.; Gérard, N.; Hulin, P.; Velazquez, L.; Turner, C.E.; Charreau, B. LNK (SH2B3) Is a Key Regulator of Integrin Signaling in Endothelial Cells and Targets α-Parvin to Control Cell Adhesion and Migration. FASEB J. 2012, 26, 2592–2606. [Google Scholar] [CrossRef]
- Kwon, S.M.; Suzuki, T.; Kawamoto, A.; Ii, M.; Eguchi, M.; Akimaru, H.; Wada, M.; Matsumoto, T.; Masuda, H.; Nakagawa, Y.; et al. Pivotal Role of LNK Adaptor Protein in Endothelial Progenitor Cell Biology for Vascular Regeneration. Circ. Res. 2009, 104, 969–977. [Google Scholar] [CrossRef]
- Wang, T.C.; Chiu, H.; Chang, Y.J.; Hsu, T.Y.; Chiu, I.M.; Chen, L. The Adaptor Protein SH2B3 (Lnk) Negatively Regulates Neurite Outgrowth of PC12 Cells and Cortical Neurons. PLoS ONE 2011, 6, e26433. [Google Scholar] [CrossRef]
- Ahlenius, H.; Devaraju, K.; Monni, E.; Oki, K.; Wattananit, S.; Darsalia, V.; Iosif, R.E.; Torper, O.; Wood, J.C.; Braun, S.; et al. Adaptor Protein LNK Is a Negative Regulator of Brain Neural Stem Cell Proliferation after Stroke. J. Neurosci. 2012, 32, 5151–5164. [Google Scholar] [CrossRef]
- Zhu, X.; Fang, J.; Jiang, D.S.; Zhang, P.; Zhao, G.N.; Zhu, X.; Yang, L.; Wei, X.; Li, H. Exacerbating Pressure Overload-Induced Cardiac Hypertrophy: Novel Role of Adaptor Molecule Src Homology 2-B3. Hypertension 2015, 66, 571–581. [Google Scholar] [CrossRef]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.-J.; Velazquez, L. The Role of LNK/SH2B3 Genetic Alterations in Myeloproliferative Neoplasms and Other Hematological Disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Gueller, S.; Nowak, V.; Sohn, J.; Hofmann, W.K.; Koeffler, H.P. Expression of the Adaptor Protein Lnk in Leukemia Cells. Exp. Hematol. 2009, 37, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Gu, Y.; Xiao, L.; Han, Q.; Li, J.; Chen, B.; Yu, J.; Kawasawa, Y.I.; Payne, K.J.; Dovat, S.; et al. Co-Existence of IL7R High and SH2B3 Low Expression Distinguishes a Novel High-Risk Acute Lymphoblastic Leukemia with Ikaros Dysfunction. Oncotarget 2016, 7, 46014–46027. [Google Scholar] [CrossRef]
- Yano, M.; Imamura, T.; Asai, D.; Deguchi, T.; Hashii, Y.; Endo, M.; Sato, A.; Kawasaki, H.; Kosaka, Y.; Kato, K.; et al. Clinical Significance of SH2B3 (LNK) Expression in Paediatric B-Cell Precursor Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2018, 183, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.-W.; Sun, Q.-Y.; Edwards, J.J.; Fernández, L.T.; Ran, X.B.; Zhou, S.Q.; Scolyer, R.A.; Wilmott, J.S.; Thompson, J.F.; Doan, N.; et al. LNK Suppresses Interferon Signaling in Melanoma. Nat. Commun. 2019, 10, 2230–2243. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.M.; Chen, X.; Qi, X.; Wang, X.M.; Li, C.Y.; Qin, R.J.; Wang, S.Q.; Liang, J.; Zeng, M.S.; Sun, C.Z. Adaptor Protein LNK Promotes Anaplastic Thyroid Carcinoma Cell Growth Via 14-3-3 ϵ/γ Binding. Cancer Cell. Int. 2020, 20, 11. [Google Scholar] [CrossRef]
- Cai, S.; Lu, J.X.; Wang, Y.P.; Shi, C.J.; Yuan, T.; Wang, X.P. SH2B3, Transcribed by STAT1, Promotes Glioblastoma Progression through Transducing IL-6/gp130 Signaling to Activate STAT3 Signaling. Front. Cell. Dev. Biol. 2021, 9, 606527. [Google Scholar] [CrossRef]
- Ding, L.-W.; Sun, Q.-Y.; Lin, D.-C.; Chien, W.; Hattori, N.; Dong, X.-M.; Gery, S.; Garg, M.; Doan, N.B.; Said, J.W.; et al. LNK (SH2B3): Paradoxical Effects in Ovarian Cancer. Oncogene 2014, 34, 1463–1474. [Google Scholar] [CrossRef]
- Pan, J.; Peng, R.; Cheng, N.; Chen, F.; Gao, B. LNK Protein: Low Expression in Human Colorectal Carcinoma and Relationship with Tumor Invasion. Biomed. Pharmacother. 2020, 121, 109467–109472. [Google Scholar] [CrossRef]
- Wang, L.N.; Zhang, Z.T.; Wang, L.; Wei, H.X.; Zhang, T.; Zhang, L.M.; Lin, H.; Zhang, H.; Wang, S.Q. TGF-β1/SH2B3 Axis Regulates Anoikis Resistance and EMT of Lung Cancer Cells by Modulating JAK2/STAT3 and SHP2/Grb2 Signaling Pathways. Cell Death Dis. 2022, 13, 472–484. [Google Scholar] [CrossRef]
- Hung, R.J.; Ulrich, C.M.; Goode, E.L.; Brhane, Y.; Muir, K.; Chan, A.T.; Marchand, L.L.; Schildkraut, J.; Witte, J.S.; Eeles, R.; et al. Cross Cancer Genomic Investigation of Inflammation Pathway for Five Common Cancers: Lung, Ovary, Prostate, Breast, and Colorectal Cancer. J. Natl. Cancer Inst. 2015, 107, djv246. [Google Scholar] [CrossRef]
- Kuo, C.-L.; Joaquim, M.; Kuchel, G.A.; Ferrucci, L.; Harries, L.W.; Pilling, L.C.; Melzer, D. The Longevity-Associated SH2B3 (LNK) Genetic Variant: Selected Aging Phenotypes in 379,758 Subjects. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Hank, S.; Dale, B.L.; Himmel, L.; Zhong, X.; Smart, C.D.; Fehrenbach, D.J.; Chen, Y.; Prabakaran, N.; Tirado, B.; et al. A Single Nucleotide Polymorphism in SH2B3/LNK Promotes Hypertension Development and Renal Damage. Circ. Res. 2022, 131, 731–747. [Google Scholar] [CrossRef]
- Lv, J.; Yu, W.; Zhang, Y.; Cao, X.; Han, L.; Hu, H.; Wang, C. LNK Promotes the Growth and Metastasis of Triple Negative Breast Cancer via Activating JAK/STAT3 and ERK1/2 Pathway. Cancer Cell Int. 2020, 20, 124. [Google Scholar] [CrossRef]
- Balko, J.M.; Schwarz, L.J.; Luo, N.; Estrada, M.V.; Giltnane, J.M.; Dávila-González, D.; Wang, K.; Sánchez, V.; Dean, P.T.; Combs, S.E.; et al. Triple-Negative Breast Cancers with Amplification of JAK2 at the 9p24 Locus Demonstrate JAK2-Specific Dependence. Sci. Transl. Med. 2016, 8, 334ra53. [Google Scholar] [CrossRef]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef]
- Mei, J.; Liu, Y.; Xu, R.; Hao, L.; Qin, A.; Chu, C.; Zhu, Y.; Liu, X. Characterization of the Expression and Prognostic Value of 14-3-3 Isoforms in Breast Cancer. Aging 2020, 12, 19597–19617. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.; Jang, S.W.; Ko, J. The Role of 14-3-3-β in Transcriptional Activation of Estrogen Receptor α and Its Involvement in Prolifertion of Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2001, 414, 199–204. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Sign. 2013, 6, 11. [Google Scholar] [CrossRef]
- Fan, X.; Cui, L.; Song, W.; Gaur, U.; Yang, M. 14-3-3 Proteins on the Crossroads of Cancer, Aging, and Age-Related Neurodegenerative Diseases. In. J. Mol. Sci. 2019, 20, 3518. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Weinstein, J. N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.-C.; Massard, C.; Lévy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef]
- Available online: www.mbcproject.org/data-release (accessed on 28 July 2023).
- Pareja, F.; Brown, D.N.; Lee, J.Y.; Paula, A.D.C.; Selenica, P.; Bi, R.; Geyer, F.C.; Gazzo, A.; da Silva, E.M.; Vahdatinia, M.; et al. Whole-Exome Sequencing Analysis of the Progression from Non–Low-Grade Ductal Carcinoma In Situ to Invasive Ductal Carcinoma. Clin. Cancer Res. 2020, 26, 3682–3693. [Google Scholar] [CrossRef]
- Kan, Z.; Ding, Y.; Kim, J.; Jung, H.H.; Chung, W.; Lal, S.; Cho, S.; Fernandez-Banet, J.; Lee, S.K.; Kim, S.W.; et al. Multi-omics profiling of younger Asian breast cancers reveals distinctive molecular signatures. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183, 1436–1456. [Google Scholar] [CrossRef]
- da Silva, E.M.; Selenica, P.; Vahdatinia, M.; Pareja, F.; Paula, A.D.C.; Ferrando, L.; Gazzo, A.M.; Dopeso, H.; Ross, D.S.; Bakhteri, A.; et al. TERT promoter hotspot mutations and gene amplification in metaplastic breast cancer. NPJ Breast Cancer 2021, 7, 1–8. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Dickler, M.N.; Shah, P.D.; Toy, W.; Brown, D.N.; Won, H.H.; Li, B.T.; Shen, R.; Vasan, N.; Modi, S.; et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat. Cancer 2020, 1, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Nixon, M.J.; Formisano, L.; Mayer, I.A.; Estrada, M.V.; González-Ericsson, P.I.; Isakoff, S.J.; Forero-Torres, A.; Won, H.; Sanders, M.E.; Solit, D.B.; et al. PIK3CA and MAP3K1 alterations imply luminal A status and are associated with clinical benefit from pan-PI3K inhibitor buparlisib and letrozole in ER+ metastatic breast cancer. NPJ Breast Cancer 2019, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Molecular Subtyping Classification | Immunohistochemical-Based Analysis | Genetic Modifications | Frequency | Treatment |
|---|---|---|---|---|
| [22] | [25] | [22] | [23,24,25,26] | [24,25,26] |
| Luminal-A | ER+, PR ≥ 20%, HER2−, Ki67 < 20% | Alterations in gene expression: ESR1, GATA3, FOXA1, XBP1. | ~75% | Hormonal therapy |
| Luminal-B | ER+, PR < 20%, HER2+, Ki67 ≥ 20% | Gene mutations: PIK3CA, ESR1, ERBB2, ERBB3. | Hormonal therapy Chemotherapy | |
| HER2-enriched | ER−, PR−, HER2+ | Gene amplifications: ERBB2, GRB7, TOPO2, MYC Gene mutations: PIK3CA. | 15–20% | Targeted therapy (anti-HER2 antibodies) |
| Basal-like | ER−, PR−, HER2− | Gene mutations: TP53, BRCA, genetic instability. | 10–20% | Chemotherapy (specific therapies are not available) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Mejía, J.A.; Mantilla-Ollarves, J.C.; Rocha-Zavaleta, L. Modulation of JAK-STAT Signaling by LNK: A Forgotten Oncogenic Pathway in Hormone Receptor-Positive Breast Cancer. Int. J. Mol. Sci. 2023, 24, 14777. https://doi.org/10.3390/ijms241914777
López-Mejía JA, Mantilla-Ollarves JC, Rocha-Zavaleta L. Modulation of JAK-STAT Signaling by LNK: A Forgotten Oncogenic Pathway in Hormone Receptor-Positive Breast Cancer. International Journal of Molecular Sciences. 2023; 24(19):14777. https://doi.org/10.3390/ijms241914777
Chicago/Turabian StyleLópez-Mejía, José A., Jessica C. Mantilla-Ollarves, and Leticia Rocha-Zavaleta. 2023. "Modulation of JAK-STAT Signaling by LNK: A Forgotten Oncogenic Pathway in Hormone Receptor-Positive Breast Cancer" International Journal of Molecular Sciences 24, no. 19: 14777. https://doi.org/10.3390/ijms241914777