
Recent Advances in Chemical Synthesis of Amino Sugars


1
Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, China
2
University of Chinese Academy of Sciences, Beijing 100049, China
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(12), 4724; https://doi.org/10.3390/molecules28124724
Submission received: 17 April 2023
/
Revised: 6 June 2023
/
Accepted: 7 June 2023
/
Published: 12 June 2023
(This article belongs to the Special Issue Carbohydrate Based Small Molecules: Sweet Spots in Medicinal Chemistry)
{kind=link}
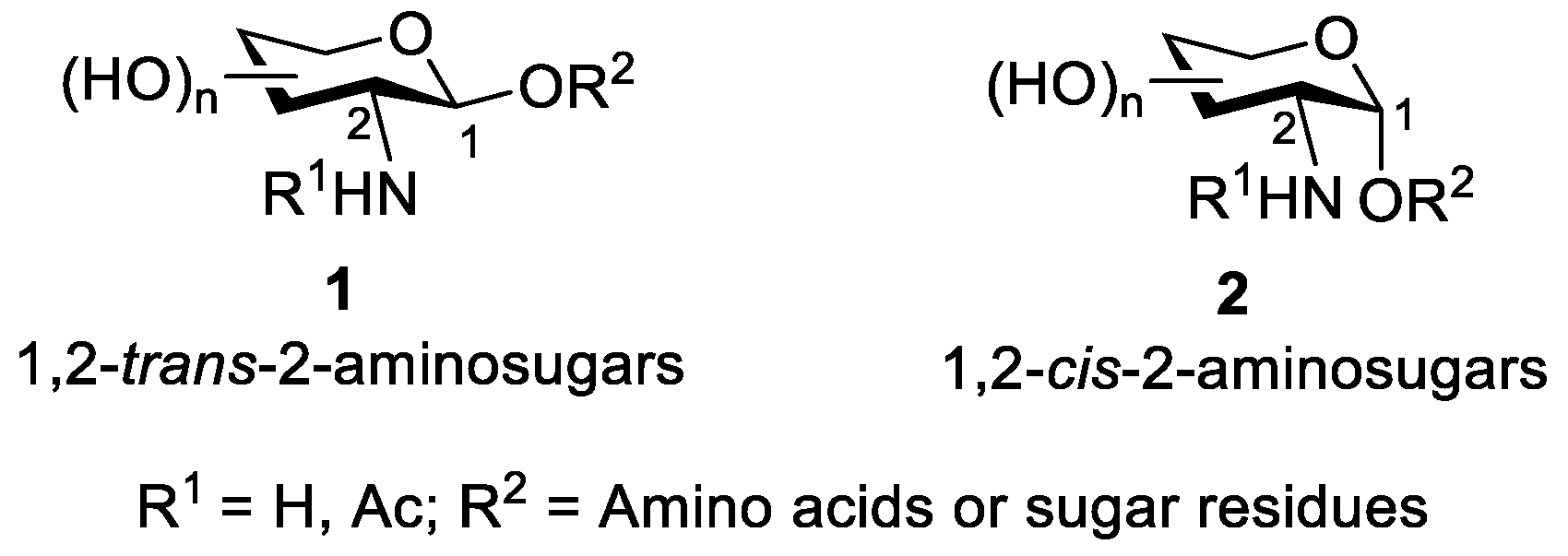{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
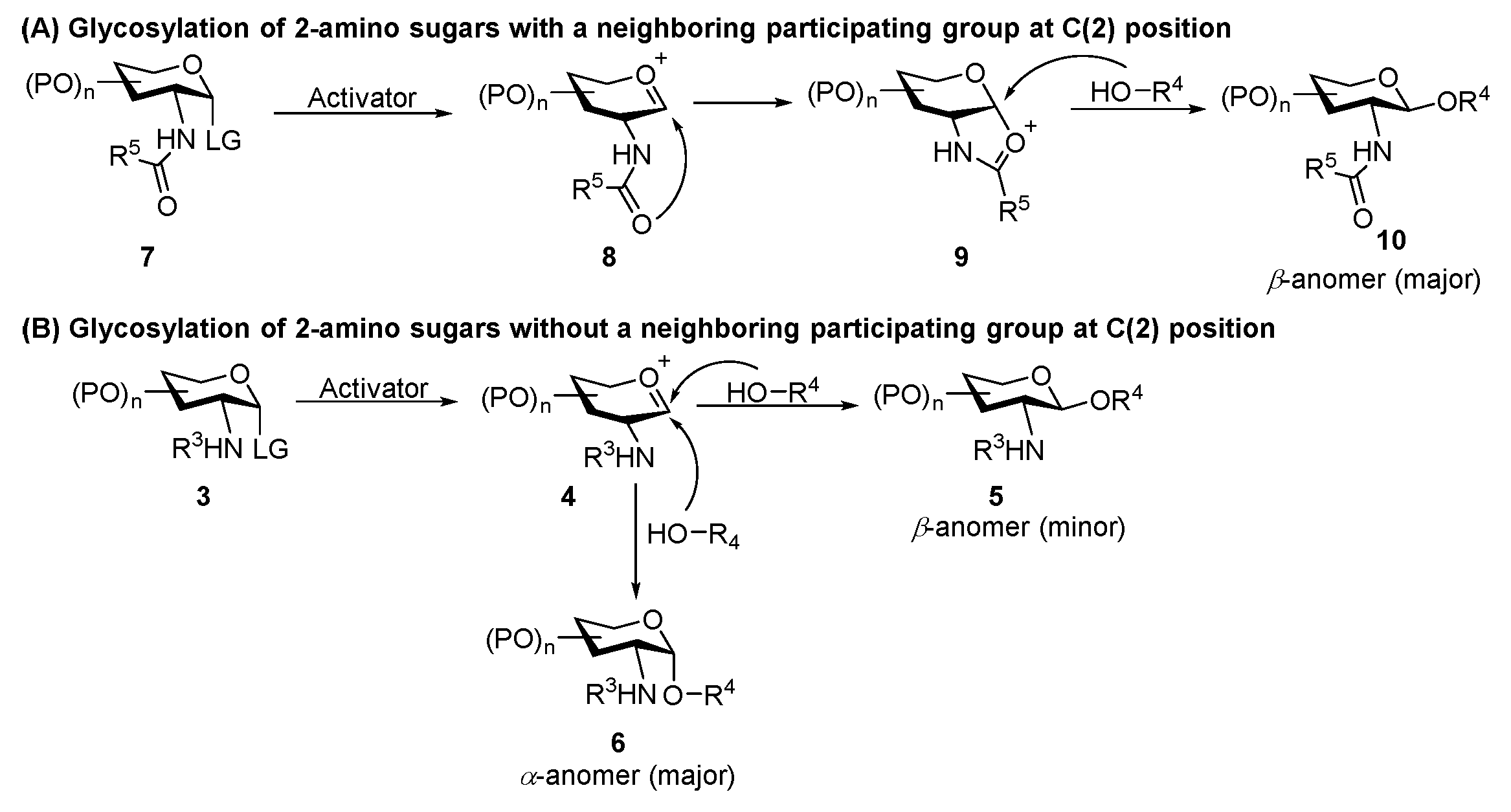{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Amino sugars are a kind of carbohydrates with one or more hydroxyl groups replaced by an amino group. They play crucial roles in a broad range of biological activities. Over the past few decades, there have been continuing efforts on the stereoselective glycosylation of amino sugars. However, the introduction of glycoside bearing basic nitrogen is challenging using conventional Lewis acid-promoted pathways owing to competitive coordination of the amine to the Lewis acid promoter. Additionally, diastereomeric mixtures of O-glycoside are often produced if aminoglycoside lack a C2 substituent. This review focuses on the updated overview of the way to stereoselective synthesis of 1,2-cis-aminoglycoside. The scope, mechanism, and the applications in the synthesis of complex glycoconjugates for the representative methodologies were also included.
1. Introduction
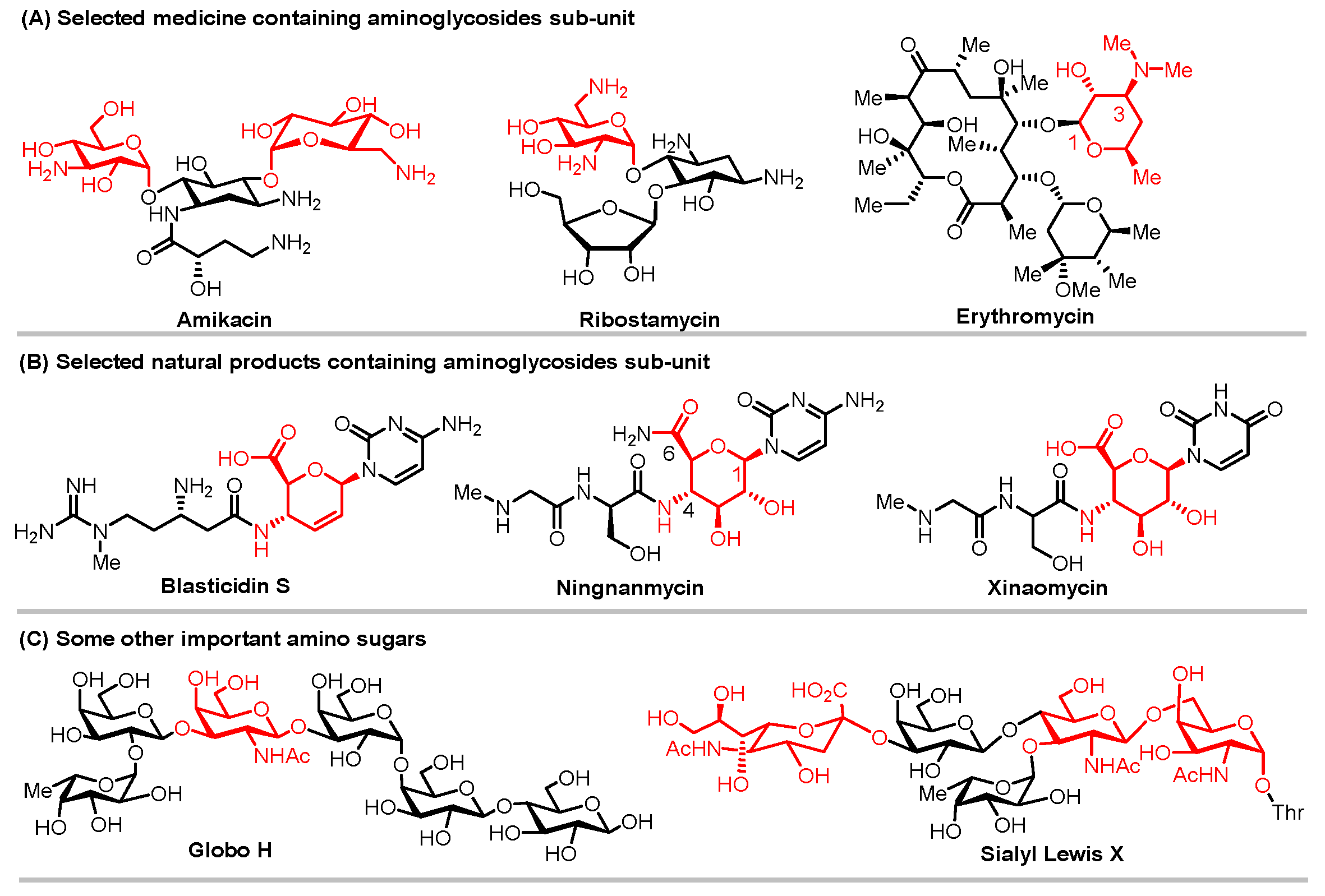
A particular kind of carbohydrates known as amino sugars have one or more hydroxyl groups replaced by amino groups. Amino sugars exist ubiquitously in nature and are present in many biologically important and naturally occurring oligosaccharides and natural products. These molecules are closely related to people’s lives and play a vital role in the human battle against diseases, and increase the quality and the yield of grain. For instance, erythromycin and other aminoglycoside and macrolide antibiotics, including amikacin and ribostamycin, which all incorporate the amino sugar sub-unit and are used as the first-line therapeutic medications for the treatment of bacterial infections, have prevented millions of deaths (Figure 1A). Amino sugars are also found in a wide range of natural products, such as Blasticidin S, Ningnanmycin, and Xinaomycin, and have been utilized successfully as agricultural antibiotics [1,2,3] (Figure 1B). Additionally, sialic acids, which are essential for cell mutual recognition, differentiation, adhesion, and malignancy, and 2-amino sugars, which are widely prevalent in various sugar complexes like polysaccharides, glycopeptides, glycolipids, and glycoproteins are both unique amino sugars. (Figure 1C) [4]. At the present stage, the availability of such compounds relies heavily on microbial fermentation, however, it is hard to quickly construct compound libraries containing multiple structural analogues in efficient ways. As a result, they are not suitable for a broad range of bioactivity screening and structure–activity relationship studies, which, ultimately, restricts the development of their activity and exploration of their functionality.
On the other hand, chemical synthesis of aminoglycosides is always associated with great challenge because: (i) the inherent regioselectivity and stereoselectivity issues in chemical synthesis of carbohydrates resulted in the lack of a general protocol to construct glycosides in an efficient manner [5,6]; (ii) competitive coordination of the basic amine to the Lewis acid promoter renders the traditional glycosylation conditions inefficient [7,8,9,10,11]; and (iii) many amino monosaccharides are not commercially available or available at a high price and often require the introduction of amino groups into the scaffolds by chemical methods through multi-step protecting/deprotecting procedures, which still encountered regio- and stereo-selective issues [12].
Despite these challenges, in view of the structural and pharmacological diversity of aminoglycosides, many elegant chemical methods were thus developed for the synthesis of aminoglycosides. In this review, we wish to give the reader an overview of the recent achievements in stereoselective synthesis of aminoglycosides by using amino sugars as glycosyl donors. Since 6-amino-6-deoxy sugars could be readily obtained from corresponding 6-OH analogues, and there are several graceful articles and reviews on the synthesis of 1-amino-1-deoxy sugars (N-glycosides) [13,14], this review does not include the synthesis of 6-amino-6-deoxy sugars and 1-amino-1-deoxy sugars, but focuses on the chemical synthesis of 2-amino-2-deoxy, 3-amino-3-deoxy, and 4-amino-4-deoxy glycosides. This mini-review is divided into the following parts: (1) glycosylation of 2-amino-2-deoxysugars with different glycosyl donors; and (2) glycosylation of 3-amino-3-deoxysugars and 4-amino-4-deoxysugars.
2. Glycosylation of 2-Amino-2-Deoxysugars
2-Amino-2-deoxysugars are commonly distributed in a wide variety of naturally occurring oligosaccharides and glycoconjugates, where they are connected to other residues via anomeric carbon to form 1,2-cis-2-aminosugars (a-anomer) or 1, 2-trans-2-aminosugars (β-anomer) [15,16] (Figure 2). In interactions between cells, many of these 2-aminosugars on cell surfaces serve as ligands for proteins, including enzymes, antibodies, and lectins [17,18,19]. With the increasing awareness of the biological importance of 2-amino-2-deoxysugars, many efficient regio- and stereo-selectively chemical methods for the synthesis of oligosaccharides containing these residues have been developed.
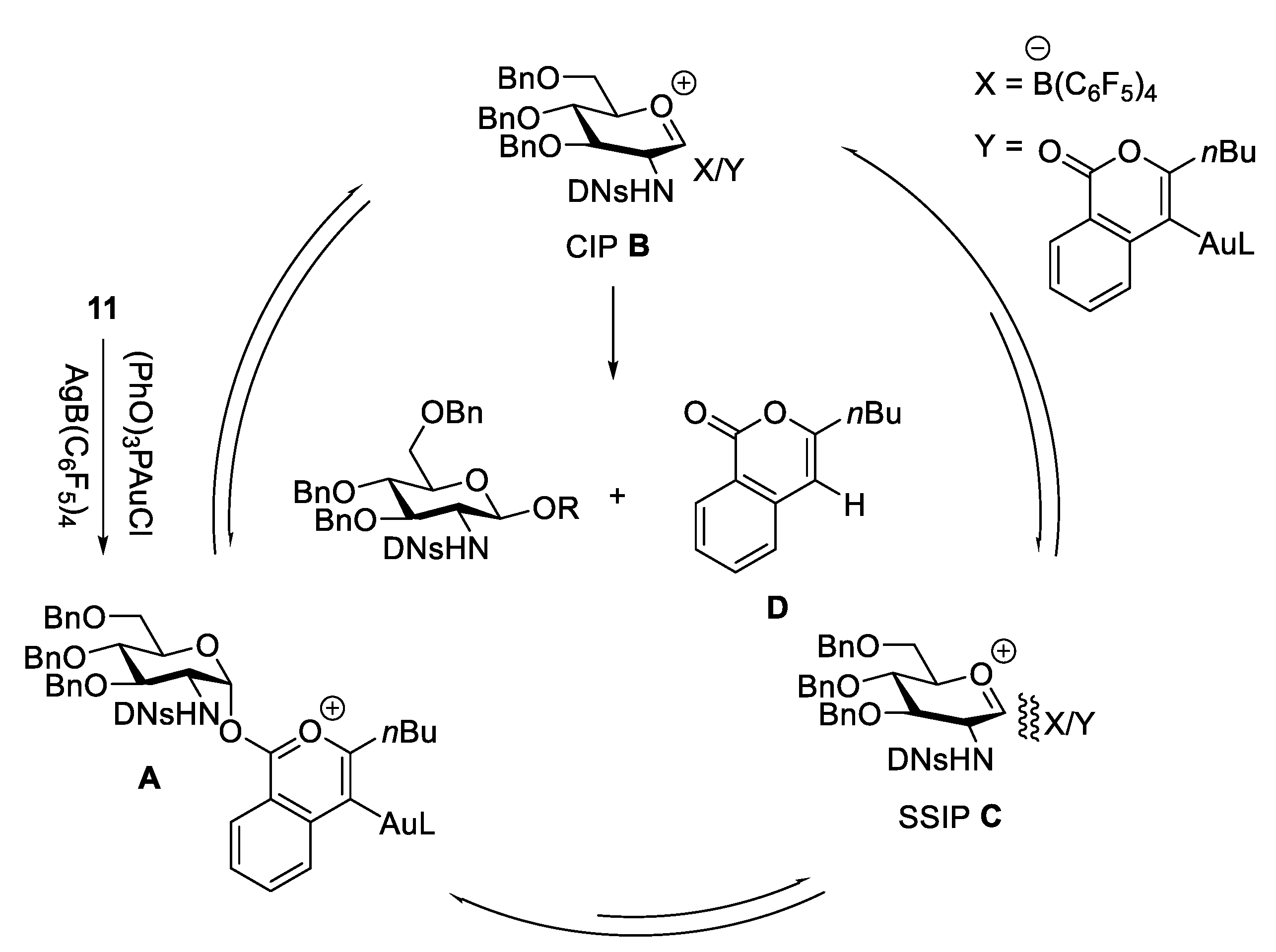
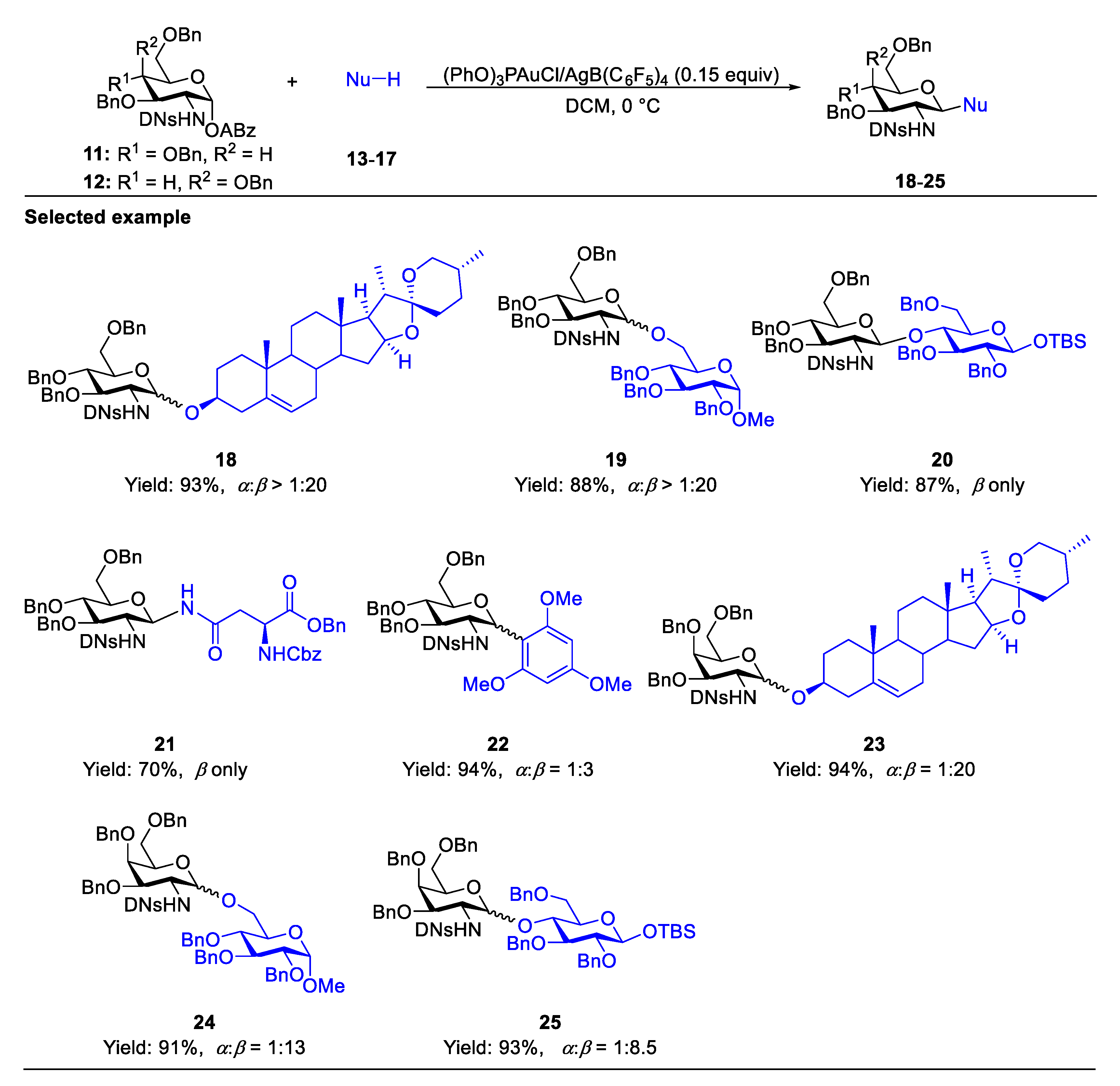
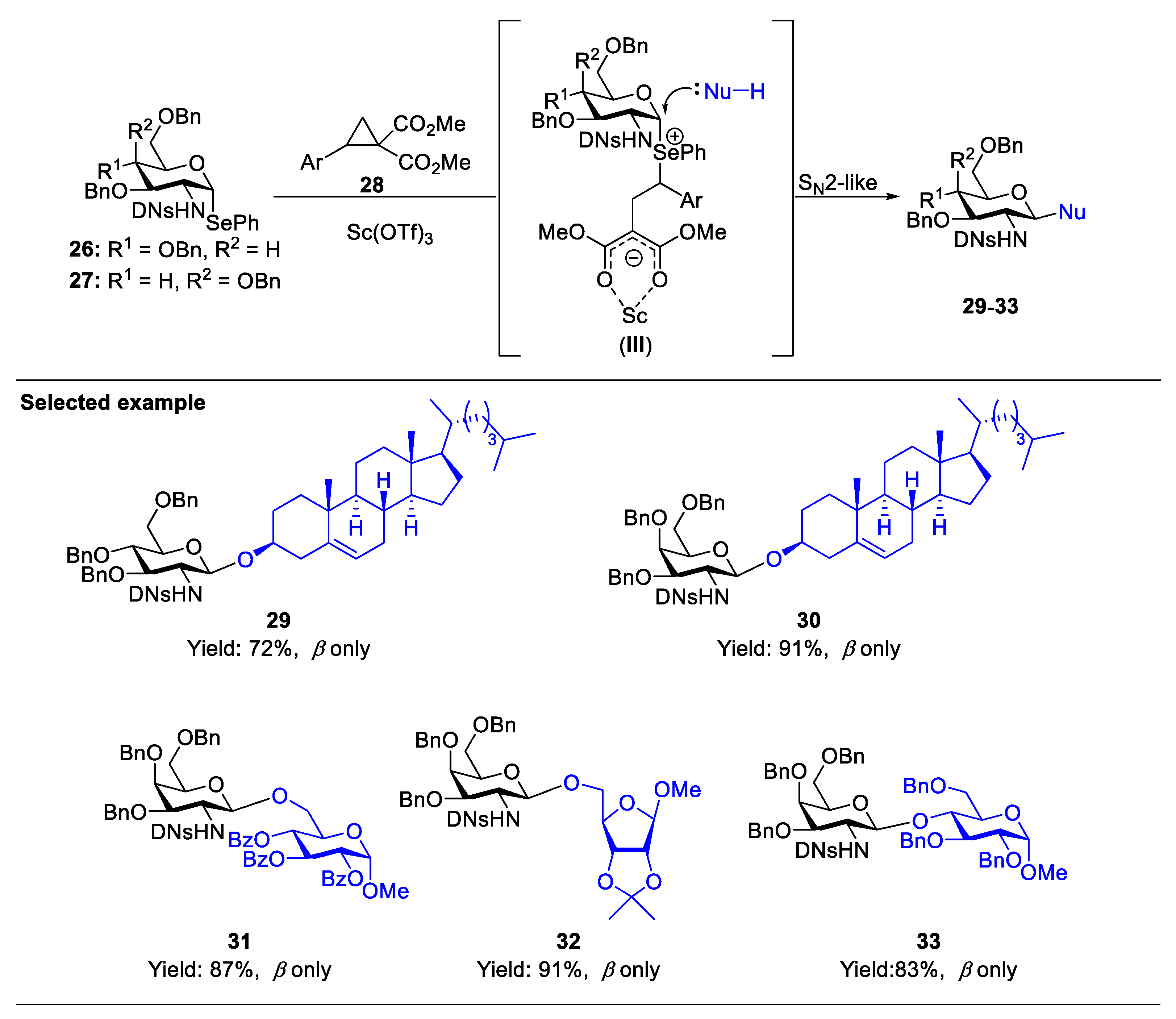
The formation of 1,2-trans-aminoglycoside which exists in nature such as chitin, chitosan, as well as many O-linked GlcNAc targets and N-linked glycans, can be reliably performed with the assistance of a neighboring participating group at C(2)-amide to provide the β-linked product 10 (Scheme 1A) [20,21,22]. There are several elegant reviews on the stereoselective formation of 1,2-trans-2-aminoglycoside [16,23,24,25]. Here, we only deal with two of the very recent representative examples. As for a non-participatory group at C(2)-amide, Li’s group developed an effective strategy for β-selective glycosylation of 2,4-dinitrobenzenesulfonyl (DNs)-protected glucosaminosyl and galactosaminosyl donors using Yu’s glycosyl o-hexynylbenzoates as the donor (Scheme 2) [26]. The methodology is compatible with a wide range of substrates, including O-, N-, and C-nucleophiles. A plausible mechanism suggested that the reaction was achieved through a SN2-like pathway. It was supposed that, compared to solvent-separated ion pair (SSIP) intermediate C, the equilibrium heavily shifts toward the intermediates glycosyloxyisochromenylium A and contact ion pair (CIP) B due to the instability of C caused by the strong electron-withdrawing effect of C(2)-DNsNH. Moreover, nucleophiles preferentially attack intermediates A and B to stereocontrollably result in β-glucosaminosides in an SN2-type process (Figure 3). Very recently, Liu and co-workers developed a donor-acceptor cyclopropane (DAC)/Sc(OTf)3 system to activate C(2)-DNsNH-protected selenoglycoside donors (Scheme 3) [27]. The mechanism suggested the DNs group would stabilize the glycosyl zwitterion intermediate (III) to engage in the SN2-like displacement to form the 1,2-trans glycosaminyl bonds in a nonparticipating manner.
1,2-cis-Aminoglycoside, the key constituents in many naturally occurring bioactive glycoconjugates, are found to have a wide range of biological activities [15,16], therefore, it is meaningful to study the corresponding synthetic methodologies. The preparation of 1,2-cis-aminoglycoside 6 is challenging as the most readily amide protecting group for amine would produce the undesired 1,2-trans-product 5 due to the participation of the neighboring-group (Scheme 1A), while a C(2)-non-participatory group would unpredictably lead to the anomeric mixtures at the newly formed glycosidic bond, even though the 1,2-cis-anomer 6 would be produced as the major product due to thermodynamic stability (Scheme 1B). Herein, we will discuss the methodologies for the construction of 1,2-cis-aminoglycoside, according to the employed glycosyl donors. The associated mechanism, the influence of additive to the stereoselectivity, and the application of the methodology to the synthesis of some biologically important oligosaccharides and glycoconjugates will also be included.
2.1. Glycosylation with C(2)-Azido Donors
The C(2)-azido functionality serves as an excellent latent amine-precursor that is stable in both acidic and basic circumstances, and can, subsequently, be selectively converted into the amino group, giving the most versatile route for the formation of 1,2-cis-aminoglycoside. Due to the chemical compatibility of C(2)-azido with many different kinds of glycosyl donors, including glycosyl halides, trichloroacetimidates, and thioglycosides, it has been applied in total syntheses of a broad range of biologically relevant oligosaccharides and glycoconjugates [28]. The needed C(2)-azide glycosyl donors could be easily prepared from the corresponding 2-amino sugars or by azidation of 2-O-triflate. The radical process from glycal was also developed by Tingoli and co-workers by using PhI(OAc)2 as radical initial and NaN3 as N3 source [29]. All of these protocols supported the application of C(2)-azido glycosyl donor for glycosylation.
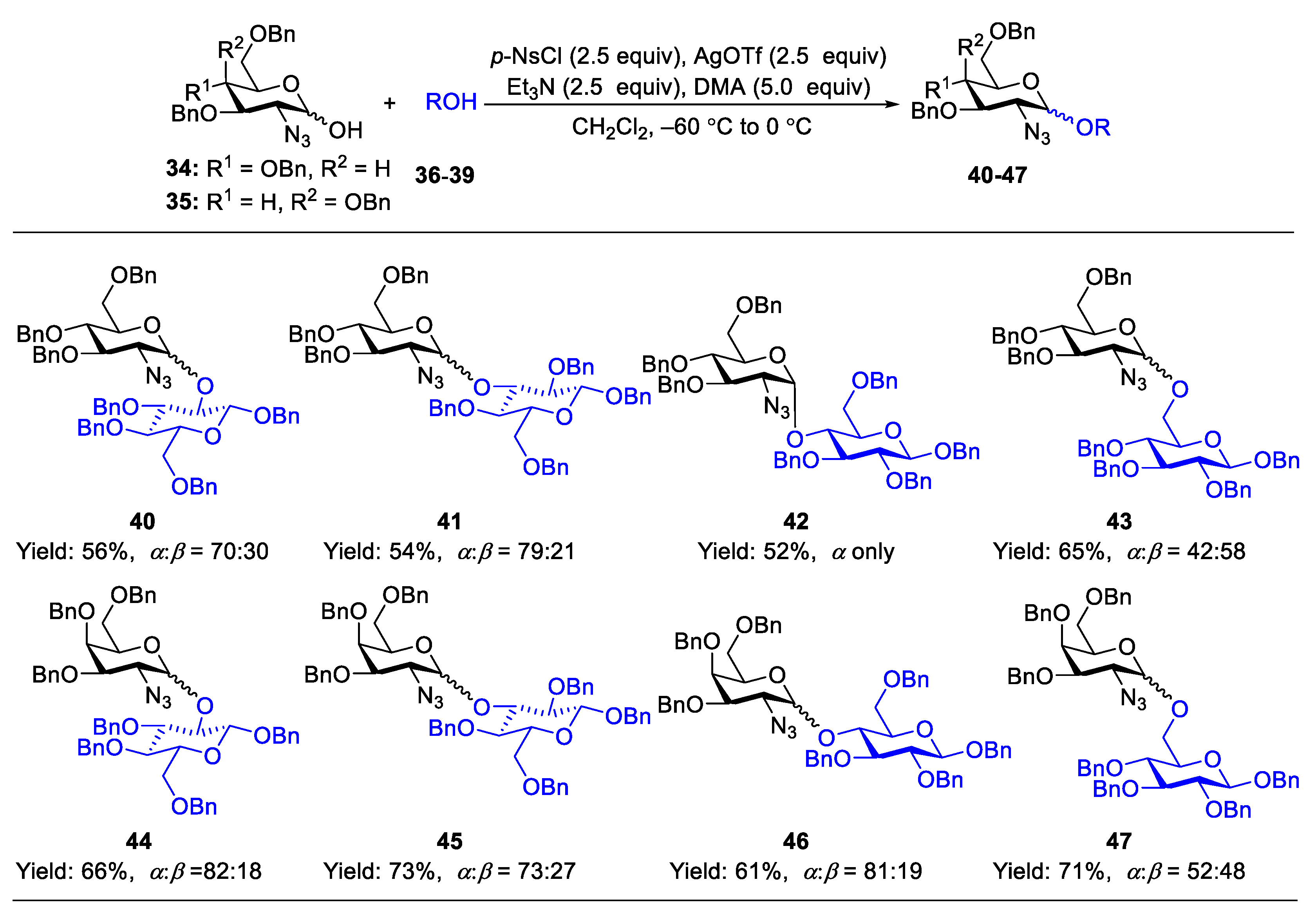
Glycosylation by in situ activation of 1-OH sugar derivatives is convenient because it avoids preparation of reactive glycosyl donors. Koto et al. reported the synthesis of α-/(1,2-cis)-glycosylation of 2-azido-3,4,6-tri-O-benzyl-2-deoxy-D-glucose and 2-azido-3,4,6-tri-O-benzyl-2-deoxy-D-galactose, which were prepared from corresponding glycals via azido-nitration followed by hydrolyzing 1-O-nitrated product on silica-gel column. By employing p-nitrobenzenesulfonyl chloride (NsCl)-silver triflate-triethylamine and N,N-dimethylacetamide (DMA) as the activating system, the anomeric free hydroxyl group could be activated in situ to produce desired products in moderate-to-good yields. The addition of DMA is crucial to control the stereoselectivity as the similar reaction in the absence of DMA lead to the formation of 1,2-trans-isomers (Scheme 4) [30].
Glycosyl nitrates are important synthetic intermediates which could be applied in subsequent functional group transformations. Demchenko’s group reported the utility of glycosyl nitrates as donors for O-glycosidation of 2-amino-2-deoxysugars in the presence of ytterbium(III) trifluoromethanesulfonate (Yb(OTf)3) (Scheme 5 and Scheme 6) [31]. Coupling the benzoylated donor 48 with acceptors 50–52 generates the desired disaccharides in 26–65% yields with good to excellent stereoselectivity (Scheme 5). In contrast to the Bz-protecting group, however, Bn-protected glycosyl nitrates delivered the corresponding products in shorter reaction time, higher yield, but worse stereoselectivity. The author concluded that this is because of the inverse correlation between the reactivity and stereoselectivity. Besides, conformation-restrainin sugar such as 2-azido-4,6-O-benzylidene-2-deoxy-3-O-triisopropylsilyl-β-D-glucosyl nitrate donor 59, was subjected to the standard reaction condition to couple with glycosyl acceptors 50–52. Despite the fact that the conformation-restraining 4,6-O-benzylidene-protected glucosyl donor produces the glycosylated product with α-selectivity [32,33,34,35], this reaction merely produced the products with lower yield and inferior selectivity (Scheme 6).
During synthesis of the core structures of glycosyl phosphatidylinositol (GPI), Guo’s group used the 2-azido-2-deoxy-D-glucopyranose donor 63 to couple with inositol acceptor 64, generating the desired product 65 and its β-isomer 65’ in 70% yield with 4:3 α-/β-selectivity (Scheme 7) [36]. The two isomers can be separated by silica gel column, facilitating its further application. Even though no detailed experimental procedure was disclosed, they used dichlorohafnocene (Cp2HfCl2) and AgOTf as promotors, which may limit its application in large-scale reactions.
In 2011, Sewald’s group reported a one-pot procedure to prepare 2-azido glycosyl chlorides via FeCl3/H2O2 mediate azidochlorination of glycals. A series of mono- and disaccharide 2-azido glycosyl chlorides were synthesized from corresponding glycals in 37–78% yields with high α-selectivity. The reaction could also be carried out at 15 g scale which produced the product in similar yield and stereoselectivity. The produced 2-azidated galactosyl donor 66 was then used to prepare glycosylated threonine building block 69 under Koenigs–Knorr glycosylation conditions. The glycosylated amino acid derivative 69 could be used as the substrate for the synthesis of TN-antigen (α-GalNAc-Ser/Thr) (Scheme 8) [37]. The yields and diastereoselectivities of the threonine glycosylation are also comparable to the corresponding bromide 67, which was obtained from an azidonitration reaction followed by bromination [38,39].
Heparin, a kind of glycosaminoglycan, is crucial to a number of important biological functions [40]. Highly selective installation of the 2-amino-α-glycosidic linkage in the heparin backbone is challenging [41]. In 2002, Seeberger’s group reported a new method for the stereoselective installation of α-glucosamine linkages by locking the conformation of the monosaccharide acceptors (Scheme 9) [42]. Coupling the donor 70 with conformation-locked glucuronic acid acceptor 71 and iduronic acid acceptor 73 provides the desired disaccharides 72 and 74 in 92% and 90%, respectively, with completely α-selectivity.
In 2007, Geert-Jan Boons and co-workers reported the glycosylation of 2-azido-2-deoxy-glycosyl trichloroacetimidates 75 and 76 with a variety of glycosyl acceptors in the presence of PhSEt or thiophene to provide disaccharides in good-to-high yields with excellent α-selectivity (Scheme 10) [43]. The enhanced α-selectivity by addition thioether was proved to be derived from successive configurational inversion via SN2-type reactions, that is, the thioether reacted with α-triflate which was confirmed by chemical shift and a smaller coupling constant, producing the β-substituted anomeric sulfonium ion then glycosyl acceptors attacked the anomeric sulfonium ion to generate the product with high α-selectivity. It was also found that in most cases: (i) elevating the reaction temperature could dramatically improve the stereoselectivity; and (ii) thiophene produced the products with better α-selectivity compared with ethyl phenyl sulfide (PhSEt). However, a similar reaction with acceptor 52 produced the product 79 in lower yield with only α-selectivity regardless of an increase in the reaction temperature or in the presence of thioether; the lower yield was because of the formation of an anomeric trichloroacetamide by-product. They also synthesized the 3,4,6-tri-O-benzyl-glucopyranosyl donor 76, and carried out glycosylation reaction under the same conditions with or without thiophene. Similar to the benzyl-protected glycosyl nitrate donors, benzyl-protected glycosyl trichloroacetimidate 76 also delivered the corresponding glycoside in lower stereoselectivity. The author speculated that the glycosylation may take place directly between acceptors with the oxacarbenium ion intermediate produced from the high reactivity of the benzyl-protected glycosyl donor.
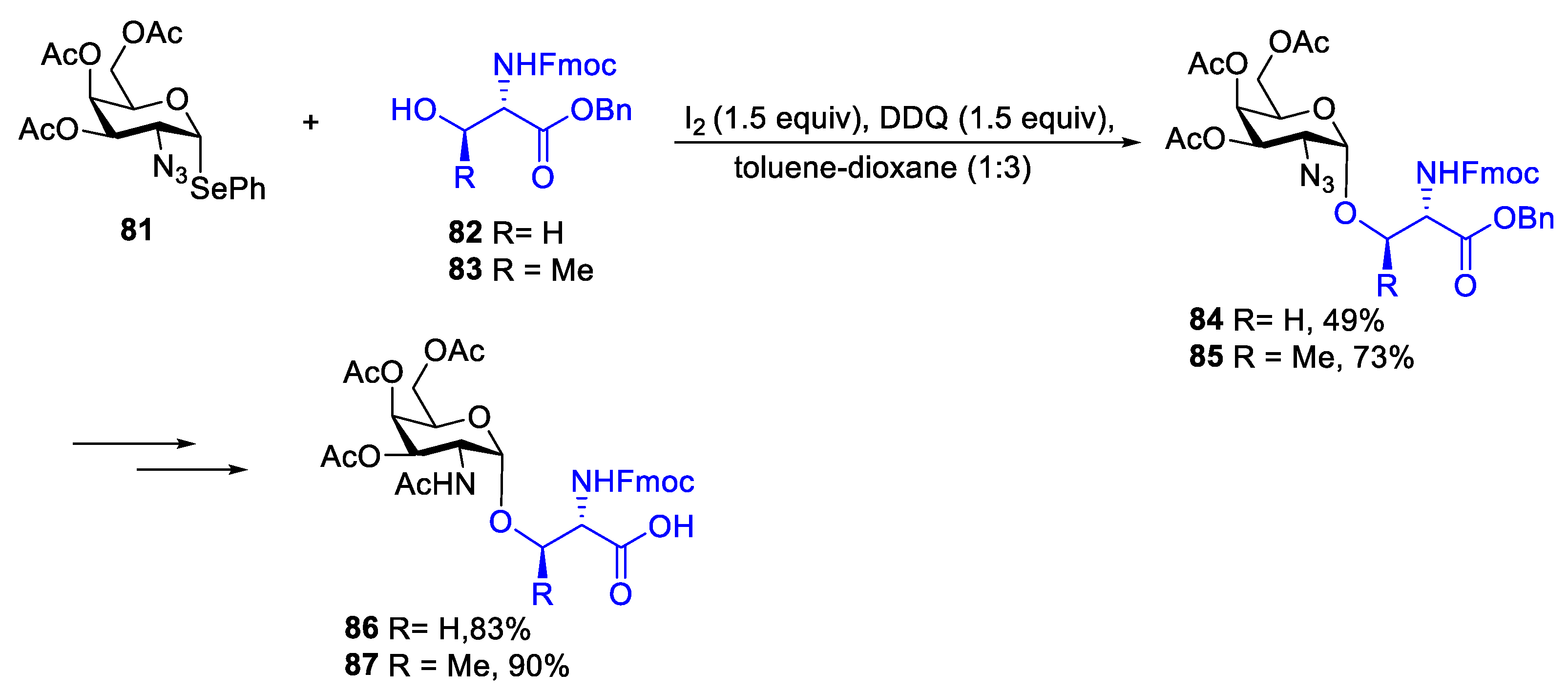
In 2008, Field’s group disclosed synthesis of mucin-related glyco-aminoacids and glycopeptides by using phenylseleno 3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-D-galactopyranoside 81 as glycosyl donor under the promotion of I2 and DDQ. In this case, selenogalactoside 81, which could be readily prepared from galactal via azidophenylselenation, was reacted with serine 82 and threonine 83 derivatives in toluene/dioxane to achieve exclusive α-stereoselectivity (Scheme 11) [44]. It was found that the use of toluene/dioxane mixture solvent and DDQ are critical for improving the yield and stereoselectivity. Even though the exact mechanism for DDQ-promoted glycosylation was not clear, they believed that high α-stereoselectivity was perhaps due to the formation of the O-β-glycosyl oxonium ion derived from dioxane.
Very recently, during the synthesis of deoxyfluorinated TN antigen analogues, Karban and co-workers prepared a series of C3 and C4 deoxyfluorinated galactosazide thiodonors to react with a variety of glycosyl acceptors and threonine derivatives in order to evaluate their stereoselectivity. It was found that for the threonine derivatives, the O-benzylated C4 fluoro-donor gave the coupling products in 63% to 69% yield with α:β selectivity ranged from 2.5:1 to 3.1:1, regardless of solvent (ether solvents such as diethyl ether and dioxane gave similar stereoselectivity as DCM alone). For C3 fluoro-donors, even though the donors with acyl and silyl protection at the 4- and 6-positions did not improve the α-selectivity, the reaction between 4,6-di-tert-butylsilylene-protected 3-fluoro galactosazide thiodonor 88 and threonine benzyl ester 83 produced the glycoside product 89 in excellent 1,2-cis-stereoselectivity (Scheme 12) [45].
Due to the protecting groups’ significant impact on the stereoselectivity of glycosyl donors, Li’s group was committed to examining the stereochemistry effect of the protecting groups [46,47,48]. They synthesized a panel of GalN3 thioglycoside donors with different protecting groups at 3-, 4-, and 6-positions, and compared the stereoselectivity of the glycosylation with primary alcohol 14. They discovered that the protecting group at 3-position had no noticeable effect on the stereoselectivity because both benzyl- and acetyl-protected thioglycoside donors delivered the products in comparable selectivity, whereas the acyl-protecting group at 4-position could increase the stereoselectivity with 4-benzoyl-protected thioglycoside producing the product in 96% yield with 1:6 of α:β. Furthermore, 2-azido-3,4,6-tri-O-acetyl-1-thio-glycoside produced the glycosylation product in a 3:1 α:β selectivity, while coupling the 6-TBDPS-protected similar donor 100 with the acceptor 14 gave the desired disaccharide product 102 in 99% yield with α:β > 20:1 [49] (Scheme 13). They also applied the optimized acyl-thioglycoside with the TBDPS protecting group at 6-position to the synthesis of precursors of TN antigen.
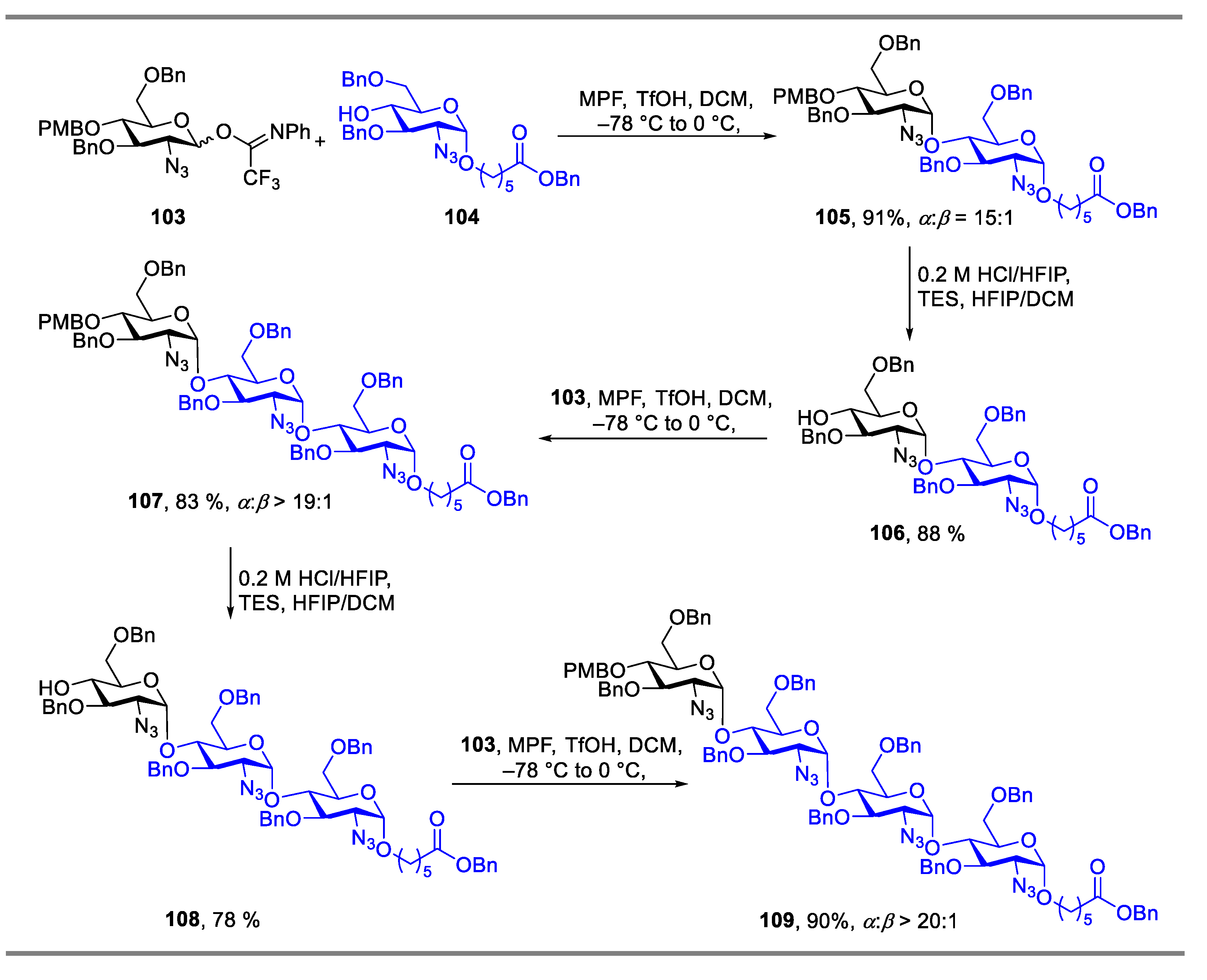
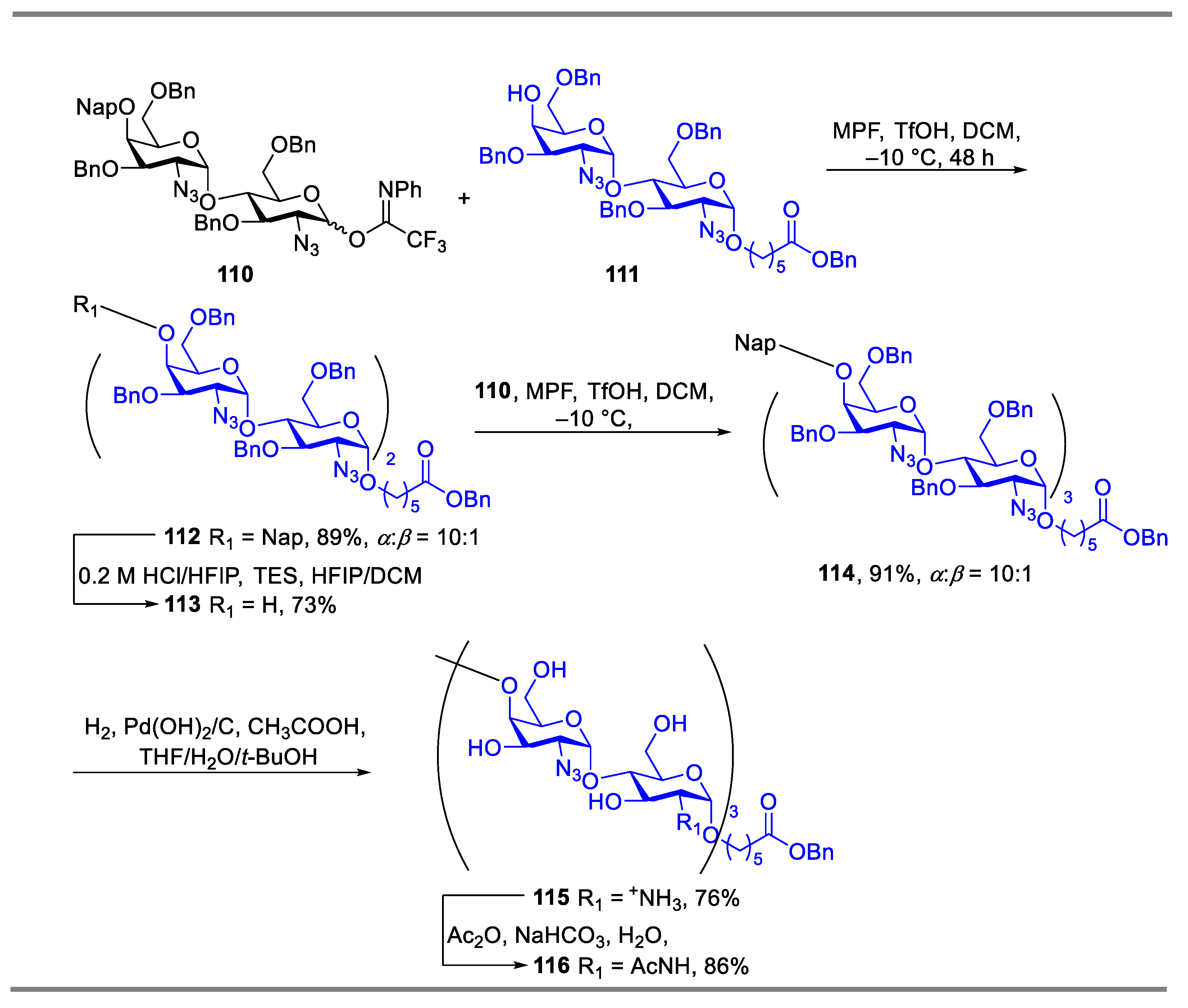
The strategy of reagent-controlled glycosylation was also employed in the synthesis of oligosaccharides. Codée’s group reported methyl(phenyl)formamide (MPF) could be employed as an additive for the glycosylation of 2-azido-2-deoxyglucose building blocks [50]. By using the strategy of MPF mediated glycosylation, an all-1,2-cis-linked tetraglucosamine was successfully synthetized. The repetitive coupling step of glycosylation reactions mediated by MPF between donor 103 and acceptor under the above identified reaction conditions provided the desired disaccharide in good yield with excellent α-selectivity (Scheme 14). Furthermore, using the same method, a fragment of Pel hexasaccharide 116 that contained both GalN and GlcN residues was also effectively synthesized in 86% yield with α:β = 10:1 (Scheme 15).
2.2. Glycosylation with 2-Nitroglycals
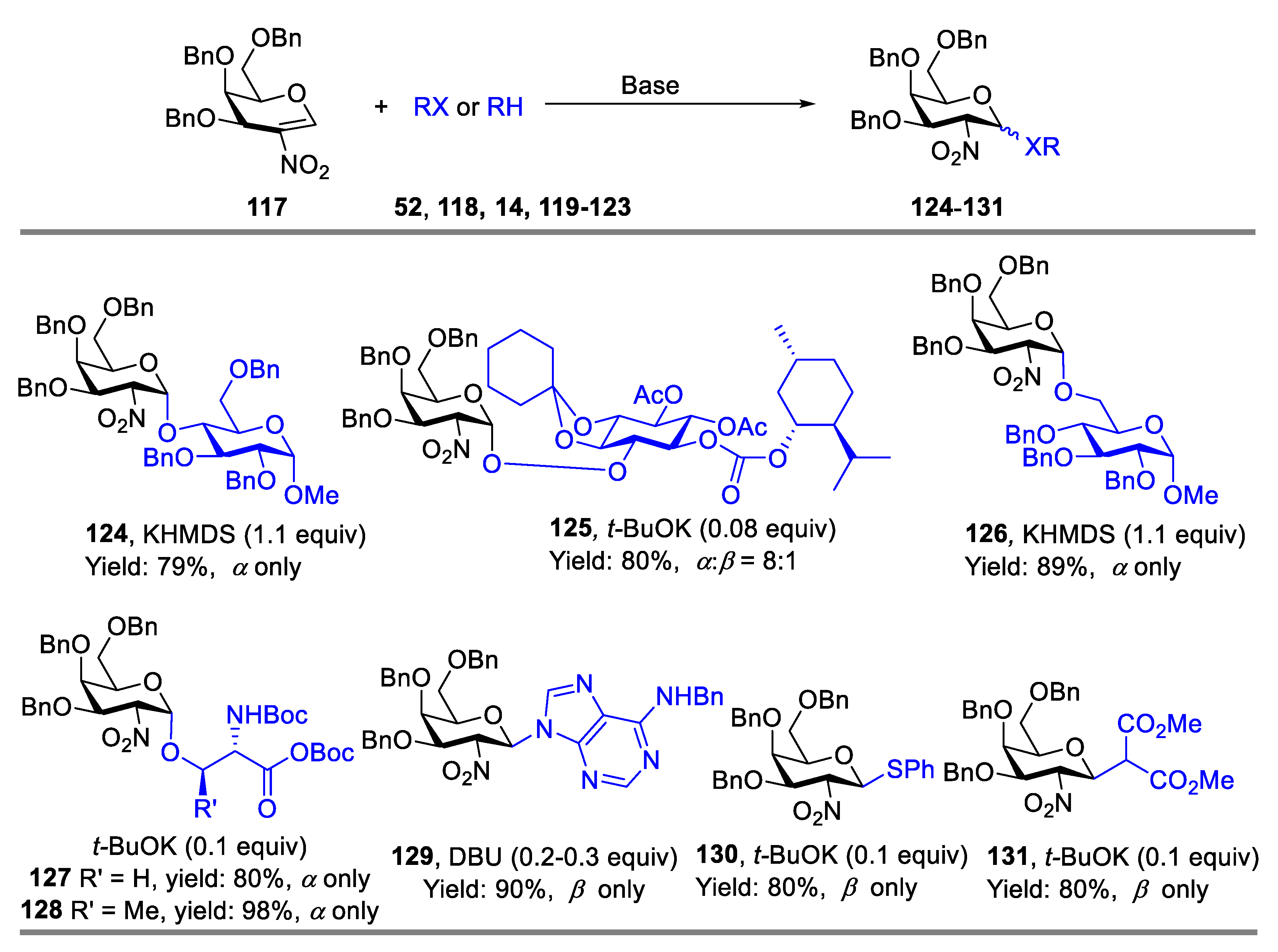
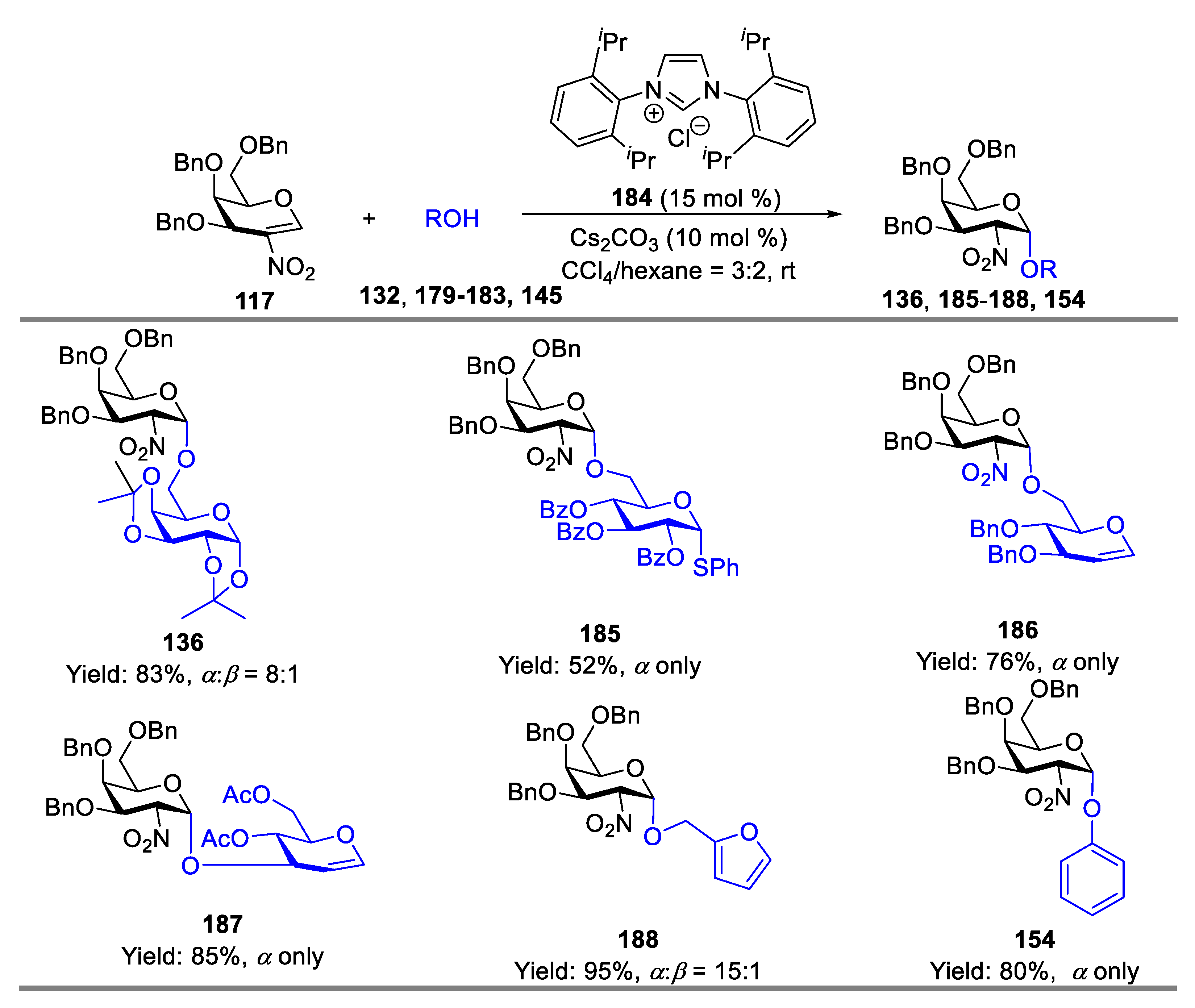
2-Nitroglycals, which could be readily prepared from corresponding glycals under nitration conditions, are important synthons in carbohydrate chemistry, and are amenable to Michael-type addition under a variety of conditions to produce 2-deoxy-2-nitroglycoside. The produced 2-deoxy-2-nitroglycoside could be readily converted into 2-amino-2-deoxyglycoside under reduction conditions. Since early work by Lemieux and co-workers [51] in the field of glycals, chemical methodologies for stereo- and regio-selective glycosylation with 2-nitroglycals have been broadly developed. Schmidt’s group has extensively studied base-catalyzed glycosylation of benzyl-protected 2-nitrogalactals with various acceptors to construct O-/S-/N-glycoside [52,53,54,55,56,57]. For instance, fully O-benzyl-protected 2-nitro-D-galactal 117 was employed by them to couple with monosaccharides, serine or threonine derivatives catalyzed by strong bases such as t-BuOK or KHMDS, providing galactosides (124–128) in high yields with good-to-excellent α-stereoselectivity, meanwhile, the same efficient glycosylation was also achieved when N,S,C-nucleophiles were used, and the corresponding N-/S-/C-glycosylation products (129–131) were produced in very good yield but with β-stereoselectivity (Scheme 16) [53,54].
Another example developed by Liu’s group using P4-t-Bu as superbase catalyst [58,59], to catalyze stereo- and regioselective glycosylation with 2-nitroglycals [60]. 2-Nitrogalactal underwent O-glycosylation with various monohydric alcohol (132 and 133) and diol (134 and 135) acceptors, providing synthetically useful 2-deoxy-2-nitro-O-glycosides in good-to-excellent yields with exclusive α-selectivity, while corresponding 2-nitroglucal gave the glycosylation products in high yield but with β:α ratio ranged from 2:1 to 1:0 (Scheme 17). The author proposed that P4-tBu reacted with alcohol (ROH) to form an ion pair (P4-t-BuH+ RO−) which facilitated alkoxide (RO−) addition to 2-nitroglycals. The stereoselectivity of the reaction is presumed because of the steric repulsion between the C4 benzyl group and the alkyl groups on ion pair (P4-t-BuH+ RO−), which repelled alkoxide-attacking 2-nitroglycals from β-face for 2-nitrogalactals and α-face for 2-nitroglucal, leading to the formation of α- and β-glycosylation products, respectively.
Due to the excellent H-bond acceptor property of nitro-group, modification of the Schmidt glycosylation protocol to construct 1,2-cis-2-aminoglycosidic linkages from 2-nitroglycal by bifunctional organocatalysts and N-heterocyclic carbene have been developed in recent years [61,62,63]. Galan’s group reported the first application of a bifunctional cinchona/thiourea organocatalyst 142 for the direct and stereoselective glycosylation of 2-nitrogalactals, affording 2-amino-2-deoxygalactosides in moderate-to-excellent yields with α-selectivity [61]. Glycosylation of 116 with glycosyl acceptors bearing electron-withdrawing protecting groups (140 and 141) generated disaccharides (143 and 144) with complete α-stereoselectivity, albeit in moderate yields (45%). Furthermore, serine 119 and threonine derivatives 120 proceeded smoothly to give the corresponding products 127 and 128 in 83% and 60% yield, respectively, and both donors’ stereoselectivity is α:β = 3:1 (Scheme 18).
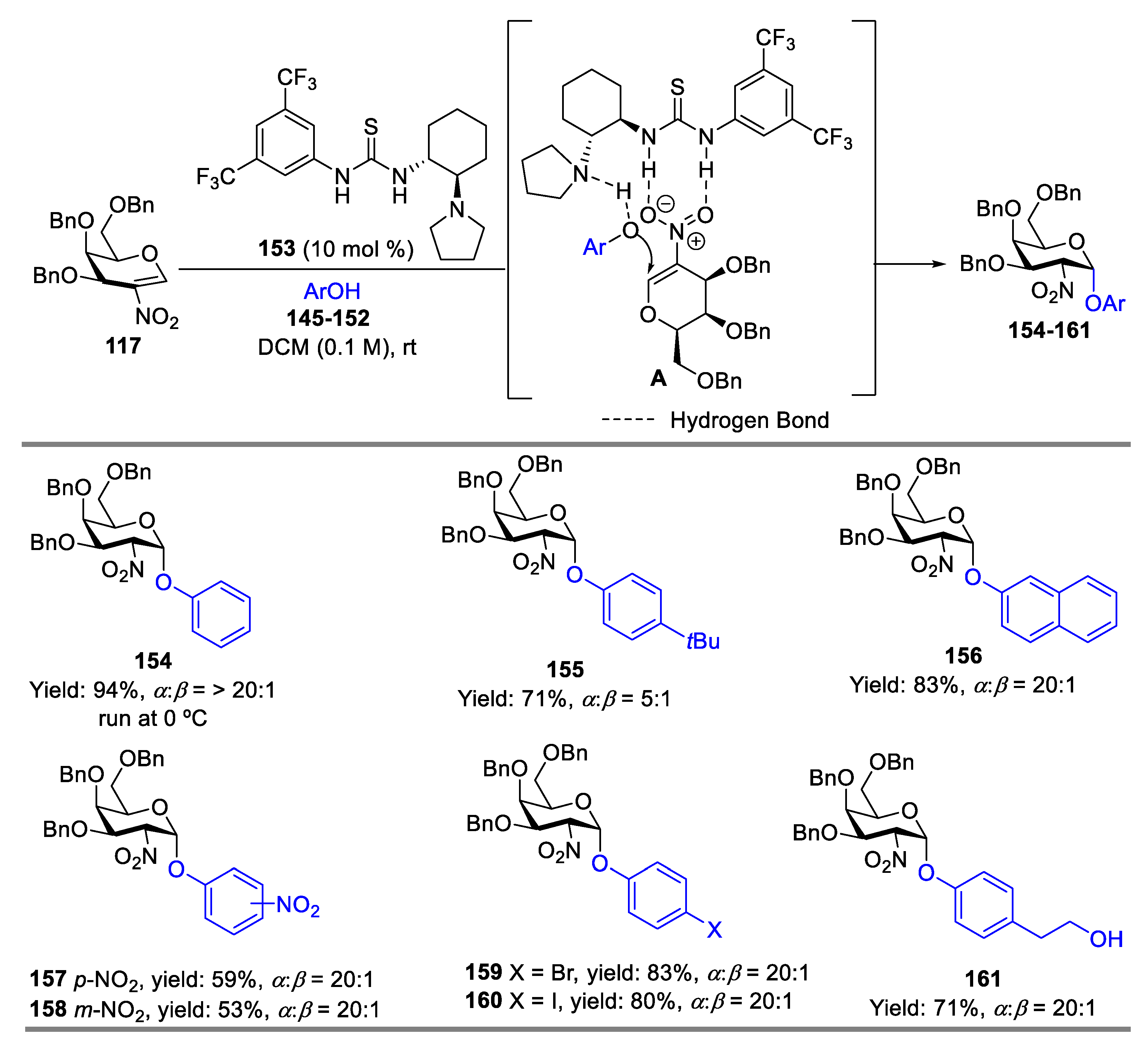
Meanwhile, Takao’s group reported an α-selective O-glycosylation procedure between 2-nitroglycals and phenol derivatives catalyzed by a bifunctional chiral thiourea catalyst 153 [63]. When 2-nitrogalactal 117 was coupled with acceptors 145–152 by thiourea catalyst at room temperature, the desired products were obtained in 53–74% yield with α:β selectivity ranged from 5:1 to 20:1 (Scheme 19). It was proved that the phenolic hydroxy groups were preferentially glycosylated even in the presence of primary alcohol to provide aromatic-O-glycosylation 161. This could be due to the basic pyrrolidine of the catalyst that assisted in the enhancement of the nucleophilic character of the phenolic hydroxyl group through phenolate formation; meanwhile, the H-bond interaction between the nitro-group in 2-nitroglycal and the thiourea catalyst charged it for the high stereoselectivity. α-Selective glycosylation of 2-nitroglucal is difficult by conventional methods; double activation by a bifunctional thiourea 153 could solve this problem, therefore, coupling 2-nitroglucal 162 with 145, 163, and 164 provides the desired products 165–167 in 75% to 95% yield with α:β selectivity ranging from 7:1 to 20:1 at −20 °C (Scheme 20).
Using conformation-constrained 2-nitroglycal as donors and a bifunctional thiourea catalyst, Sun and coworkers recently described a new protocol for the highly efficient and stereoselective production of 1,2-cis-2-amino-2-deoxy-glycosidic linkages [64]. Notably, 2-nitroglucal donors, which are prone to provide glycosylation products with lower α-stereoselectivity than their 2-nitrogalactal counterparts by using conformation-constrained strategy, delivering the disaccharides in high yield with good-to-excellent stereoselectivity, highlighting the beneficial effect of the cyclic protecting group on stereoselective glycosylation of 2-nitroglucals. For example, glycosylation of conformation-constrained 2-nitroglucal donor 168 with acceptors 77, 132, 14, and 169 in the presence of thiourea catalyst 170 provided the desired disaccharides 171–174 in 45–86% yields with α:β selectivity ranged from 2.7:1 to 20:1 (Scheme 21). Similarly, 2-nitrogalactal 175 as donor also provided α-glycoside as the major products, the cyclic di-tert-butylsilylidene group in 175 steered the glycosylation reaction to proceed with excellent α-stereoselectivity, regardless of acceptors with primary or secondary alcohols (Scheme 22).
In recent years, N-heterocyclic carbene (NHC) has emerged as a powerful organocatalyst in organic chemistry [65,66,67,68,69,70]. The key premise in NHC catalysis is the formation and the involvement of an enaminol intermediate, known as the Breslow intermediate [69]. Additionally, NHC-catalyzed reactions through noncovalent bonding interactions have also been developed [71,72,73,74]. Chen and co-workers reported the first NHC catalyzed stereoselective glycosylation of 2-nitrogalactals with alcohols and phenol [62]. A variety of functional groups in the acceptors were compatible in this approach, and the corresponding products were obtained in good-to-excellent yields (52–85%) with high-to-excellent α-selectivity (α:β = 8:1 to α only) (Scheme 23).
In 2022, Zhang and co-workers reported stereoselective synthesis of 1,1′-2-amino thiodisaccharides using 2-nitroglycals as donors catalyzed by bifunctional organothiourea [75]. Very recently, the methodology for the regio- and stereo-selective convergent synthesis of 2-amino-2-deoxy-dithioglycosides from 2-nitroglycals was also disclosed by the same group [76].
2.3. Glycosylation with Ring-Fused 2,3-Oxazolidinone Thioglycosides
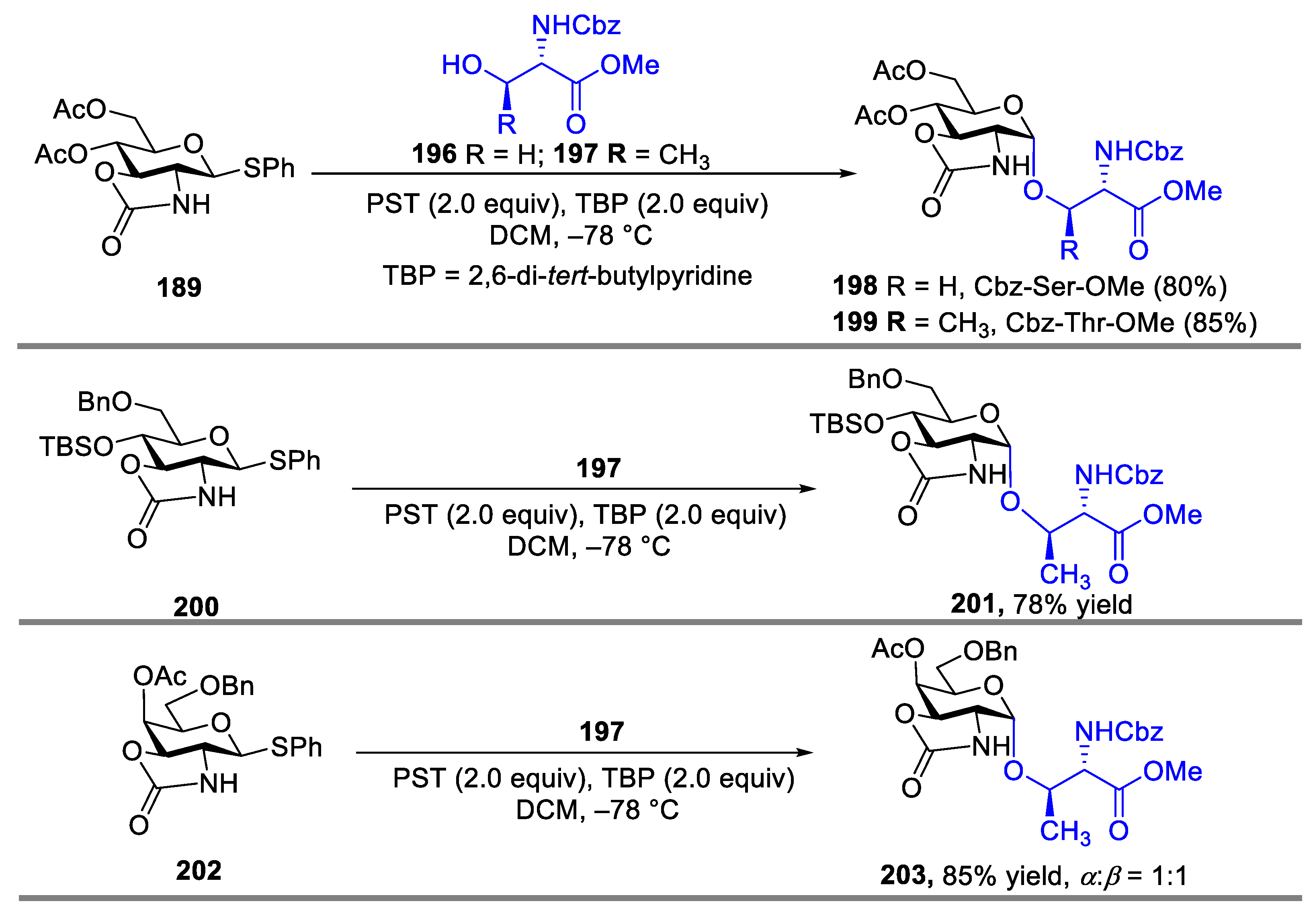
In 2001, Kerns and coworkers reported a protocol using 2,3-trans-oxazolidinone as an effective protective group with high α-selectivity toward glycosylation. A notable feature of this class of α-selective D-glucosaminyl donors is that the oxazolidinone group affords simultaneous protection of the 2-N and 3-O positions [77]. Coupling donor 189 with acceptors 190–192 in the presence of PST (phenylsulfenyltriflate, PhSOTf) and 2,6-Di-tert-butyl-4-methylpyridine (DTBMP) gives the α-linked glycoside 193–195 in 75–97% (Scheme 24).
Later, the same group expanded this methodology to the formation of α-linked serine and threonine derivatives [78]. No matter what the protective groups on the sugar were, the desired products could be obtained in high yield (78–85%) with exclusive α-selectivity (Scheme 25), however, when the similar reaction was performed with fused-ring 2,3-oxazolidinone galactosamine donor, poor stereoselectivity was observed. Even the glycosylated production was isolated in high yield. The same poor selectivity was also observed with other 2-azido-2-deoxy-D-galactosyl donors (sulfoxide, thiophenyl, and selenoglycoside donors) in the previous reports [79,80,81].
In 2004, Kerns’s group modified the oxazolidinone group to its N-acetyl analogues, leading to switch the reaction from α-selectivity to β-selectivity [82]. In Kerns’s case, stereoselective formation of α-linked or β-linked glycoside was supposed to be dependent on the reactivity and steric hindrance of acceptors. Due to the steric hindrance from the acetyl group on N-atom, and the equilibrium between α-triflate (a major product but with less reactivity) and β-triflate (a minor product but with high reactivity), the more-reactive and less-steric-hindered alcohol reacted rapidly with α-triflate to produce β-linked glycoside; conversely, less-reactive alcohols with steric hindrance could react with minority but more-reactive β-triflate to obtain α-linked glycoside with extended reaction time (Scheme 26).
In 2005, Stefan Oscarson and co-workers found that when N-acetylated 2N,3O-oxazolidinone thioglycoside was activated with NIS and AgOTf in the presence of glycosyl acceptors, the corresponding glycosylation products could be formed in excellent yield [83]. Interestingly, the stereochemistry of the anomeric carbon could be switched by controlling the amount AgOTf. When the catalytic amount of AgOTf was used, the reaction delivered the products in exclusive β-selectivity (Scheme 27, condition A), while 0.4 equiv. of AgOTf resulted in forming the products in total α-selectivity (Scheme 25, condition B). A mechanism study has shown that β-coupling products could take place in situ anomerization to form α-glycosylation products through Ag cation-promoted endocyclic C-O bond cleavage intramolecular cyclization [84].
In 2008, Ito and co-workers reported a stereoselective α-glycosylation procedure using N-benzyl-2N,3O-oxazolidinone thioglycoside as a highly α-directing glycosyl donor [85]. It was found that the glycosylation reaction between thioglycoside donors 223–224 and primary alcohols produced the products in lower selectivity, however, the stereoselectivity could be improved by using toluene and 1,4-dioxane as mixture solvent, meanwhile, a similar reaction with less-reactive secondary alcohol could produce the products in high selectivity regardless of solvents (Scheme 28). The key features of this reaction included: (i) the reaction was performed at room temperature and could be run on large scale; and (ii) the procedure could be used for the preparation of trisaccharide with two 1,2-cis-glycosidic bonds by one-pot two-step sequential glycosylation reaction (Scheme 29), which rendered it possible to use in polymer-supported or automated oligosaccharide synthesis.
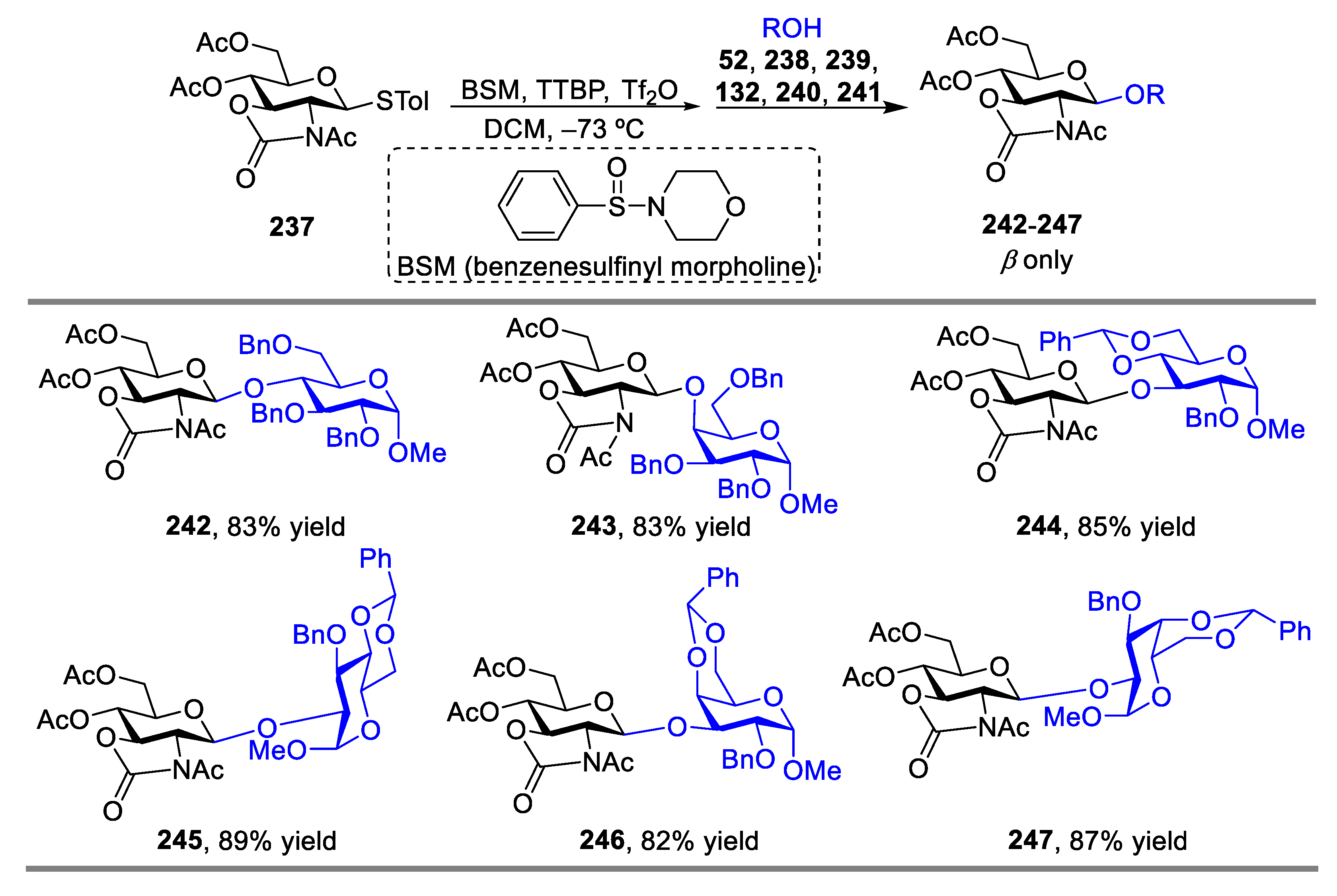
Although great advances in this field have been achieved by use of 2,3-trans-oxazolidinone as a non-participating group for glucosamine donors, several drawbacks, such as the undesired glycosylation and sulfenylation of the nitrogen atom [77,78], reduction of α-selectivity [82], low yields [85], and limited scope of acceptors [84,85], still existed. To tackle these problems, a new efficient strategy based on pre-activation protocol for both α- and β-stereoselective glycosylation of glucosamine donors was developed by Ye’s group [86]. In this case, a glycosyl donor such as 4,6-di-O-acetyl-N-acetyl-oxazolidinone protected donor 237, was completely activated and consumed first, then a glycosyl acceptor was added. By using this strategy, either excellent α-stereoselective or β-stereoselective glycosylation could be achieved in the absence of any base or by the addition of a hindered base, such as 2,4,6-tri-tert-butylpyrimidine (TTBP) [87] (Scheme 30 and Scheme 31). Compared with the traditional glycosylation protocols, controllable glycosylation could be realized by simply adding a steric-hindered base or without addition of any base under pre-activation procedure, which facilitated the construction of complex oligosaccharides with important biological functions. The same group also discovered that the protected group in the oxazolidinone donors could dramatically influence the stereoselectivities of the pre-activation procedure [88].
2.4. Glycosylation with C(2)-Benzylidenamino Donors
2.4.1. The Development of Nickel-Catalyzed Stereoselective Glycosylation
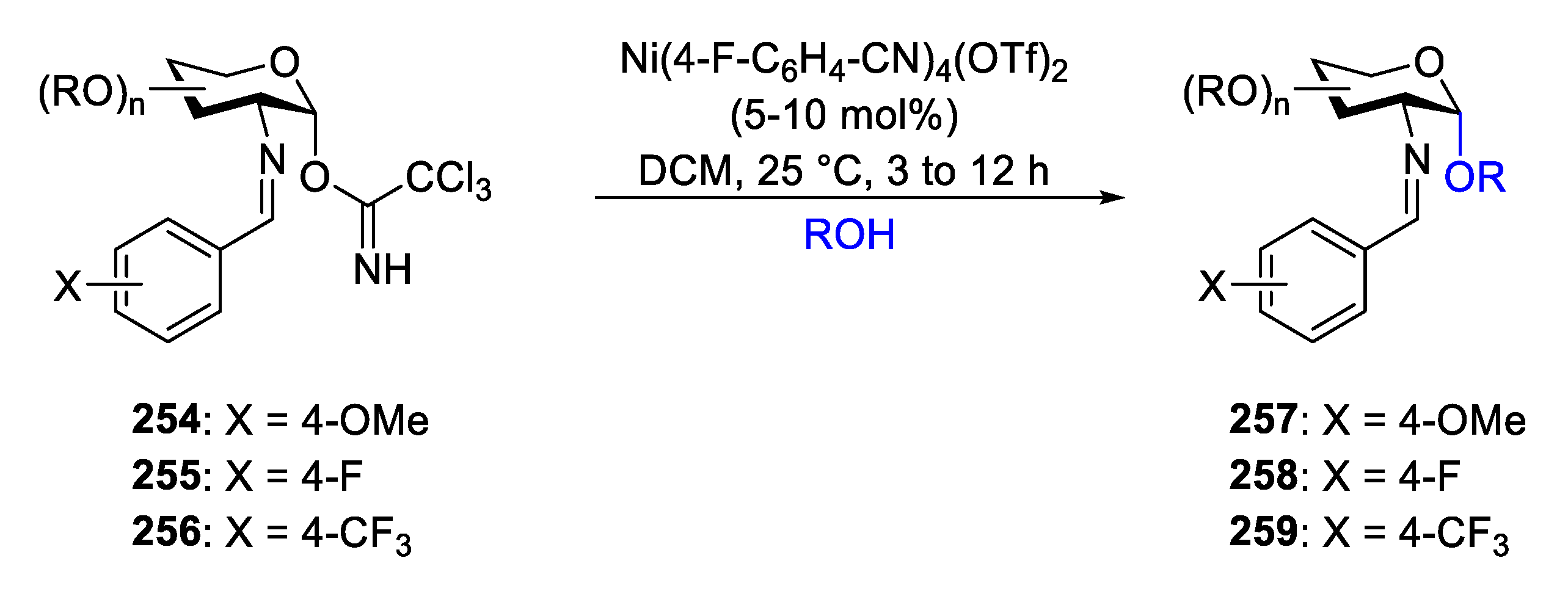
The Nguyen’s group focused on the development of novel, scalable approaches to avoid the issues associated with the formation of 1,2-cis-2-aminoglycoside by using cationic nickel (II) catalysis [89,90]. Trihaloacetimidate (TCA) is viewed as a good “leaving group” in carbohydrate chemistry whose nitrogen can be coordinated with metals to play a role as a “directing group” in transition-metal-catalyzed reactions. Additionally, the benzylidene at C(2)-amino group of sugars serves not only as a “protecting group” but also as a “directing group” in transition-metal-catalyzed reactions [91]. In 2009, The Nguyen’s group reported a nickel-catalyzed stereoselective glycosylation with C(2)-N-para-(methoxy)benzylideneamino donor 254 for the formation of 1,2-cis-2-aminoglycoside (Scheme 32) [89]. The thorough adjustment of the reaction conditions revealed that the catalyst, electronic properties of the ligands, and the substituted group on the benzylidenamino protecting-group all played crucial roles in regulating the stereoselectivity and speeding up the reaction.
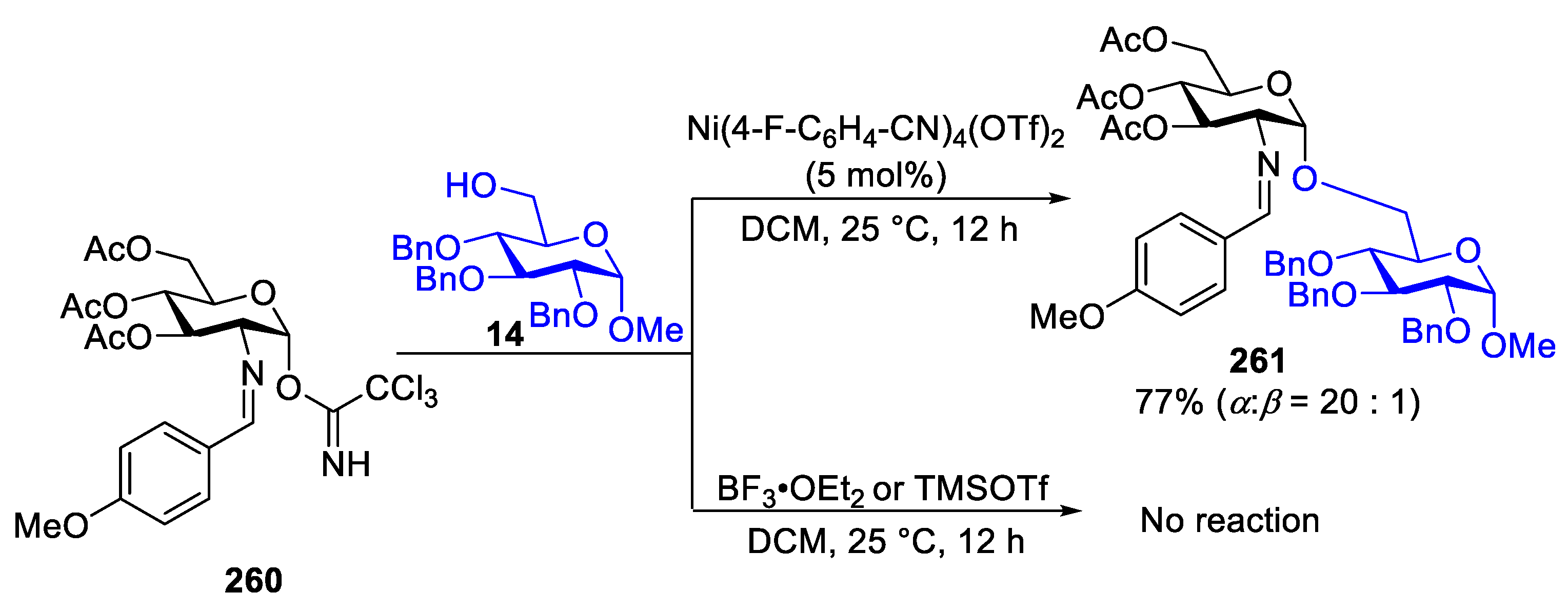
In order to prove the superiority of the catalytic system in the glycosylation reaction, comparisons against conventional trichloroacetimidate donor-activation protocol and the developed nickel-catalyzed formation of 1,2-cis-aminosugar was performed. Classical Lewis acid activator of TCA donor, such as TMSOTf and BF3·OEt2, either resulted in the decomposition of the glycosyl donor or led to the formation of a trace amount of the desired disaccharide 261 [92] (Scheme 33). Compared to these established glycosylation methods [88,92], the coupling of 260 with 14 proceeded smoothly to provide disaccharide 261 in 77% yield with excellent α-selectivity.
Glucosamine donors 260, 262–264 were then reacted with a broad range of glycosyl acceptors, such as primary, secondary, and tertiary alcohols (Scheme 34). All the glycosylations with cationic nickel(II) catalyst produced the desired products 261, 270–282 in high yield with excellent α-selectivity. The donor with steric hinderance, such as tertiary adamantol, also produced the desired product 282 in 96% yield and with α-selectivity (α:β = 17:1). Dihydrocholesterol 266, a secondary alcohol acceptor, was employed to couple with the donor 260 to produce glycoconjugate 275 in 85% yield with α-selectivity (α:β = 11:1). Increased yield and α-selectivity are observed when glucosamine donors are combined with acceptor 260 when the para-substituent on the benzylidene group is simply changed from p-OMe-benzylidene in 277 (α:β = 6:1) into p-CF3-benzylidene (α only). Meanwhile, it was found that electron-withdrawing C(2)-N-para-fluoro- and trifluoromethyl-benzylidenamino donors 263 and 264 are more stable compared to the methoxy donor 260 for purification [62], which would facilitate it for preparation on a large scale.
Additionally, galactosamine trichloroacetimidate 283 was also employed as a donor to couple with both primary and secondary alcohol acceptors (Scheme 35). Once again, these results showed the cationic nickel (II) catalyst in the glycosylation reaction could produce 1,2-cis-2-aminoglycosides in high yield with outstanding α-selectivity regardless of the kind of the acceptors.
Additionally, the nickel method was also suitable for oligosaccharide synthesis by (1 + 2), (2 + 1), or (2 + 2) process (Scheme 36). In a (1 + 2) strategy, the electron-rich disaccharide acceptors gave a higher yield than the electron-deficient one, however, the selectivity of these two acceptors is almost uniform (294 vs. 296, and 295 vs. 296). Coupling the secondary alcohol of disaccharide acceptor 292 with donor 264 provides the desired trisaccharide 298 in 76% yield with a high α-selectivity (α:β = 11:1). In a (2 + 2) strategy, coupling disaccharide acceptor 292 with disaccharide donor 289 proceeded smoothly to provide tetrasaccharide 302 in 76% yield with α-selectivity (α:β = 11:1).
Due to the lower reactivity, using D-glucuronic acid derivatives as acceptors to couple with donors in a glycosylation reaction generally gave the products in low yield and poor selectivity. However, the nickel-catalyzed strategy might be able to partially resolve this issue [93,94] (Scheme 37). For instance, coupling acceptors 303–305 with donors 260, 262–264 proceeded smoothly to provide the desired disaccharides in moderate-to-good yields (55–87%) with good α-selectivity (α:β = 9:1 to α only). Among different benzylidenamino protected donors (260, 262–264), the one with an electron-withdrawing group, such as para-(trifluoromethyl)benzyl (264), was found to be the most efficient. Glycosylation of the methyl ester of D-glucuronic acid acceptor 305 with donor 264 produced the desired disaccharide 311 in 87% yield with exclusive α-selectivity.
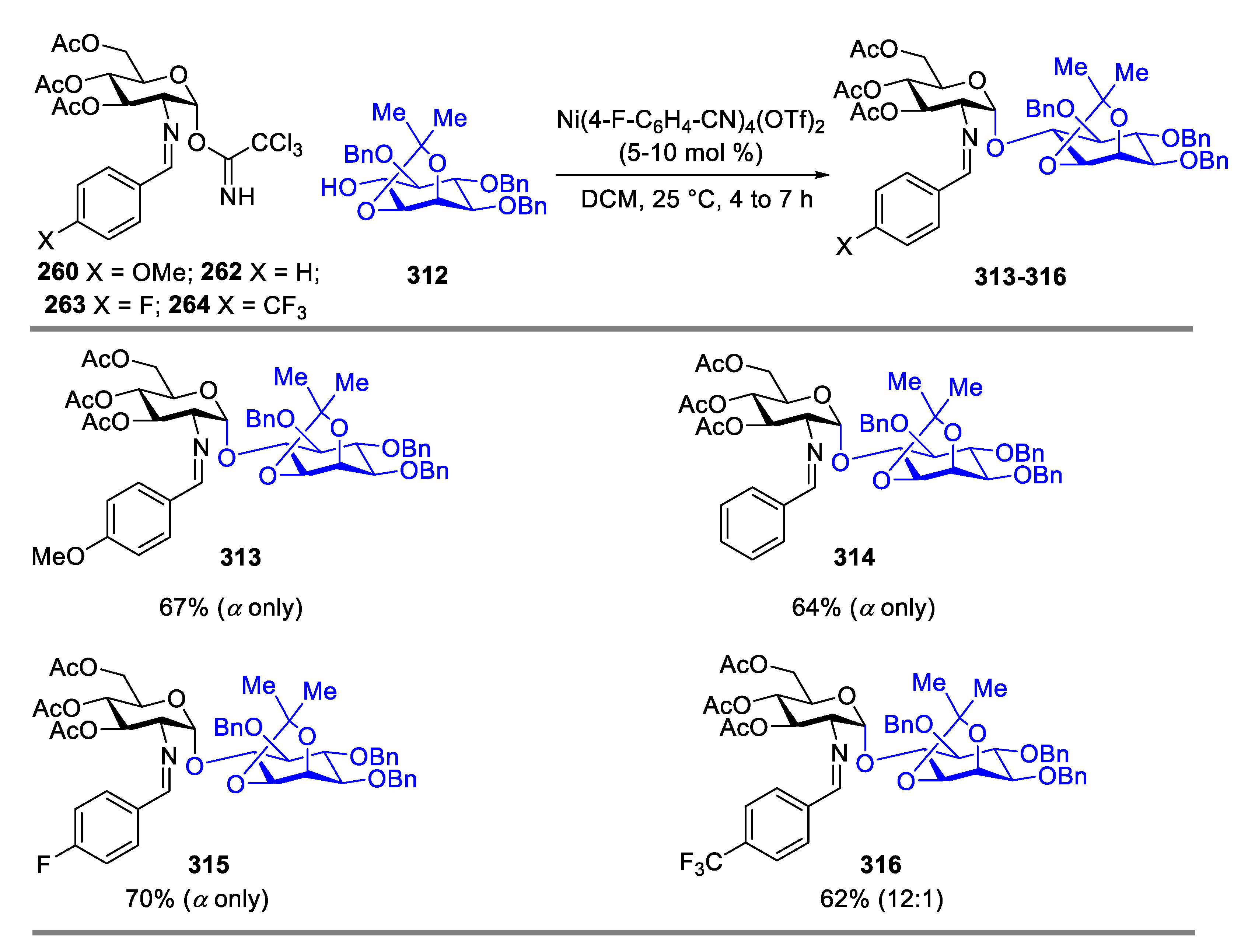
Glycosylphosphatidylinositol (GPI) anchors are a large family of glycolipids that play significant roles in various biological processes, including signal transduction, prion disease pathogenesis, and immune response [95,96,97,98,99]. Total synthesis of GPI anchors is challenging due to structural complexity. The available methods of construction of the pseudodisaccharide unit of D-glucosamine and inositol components had several limitations, such as poor α-selectivity and long reaction time [100,101,102,103,104]. Coupling inositol nucleophile 312 as the acceptor with C(2)-N-substituted benzylidene D-glucosamine TCA donors 260, 262–264 produced desired products 313–315 in excellent α-selectivity (Scheme 38), addressing the issue of poor α-selectivity in the literature.
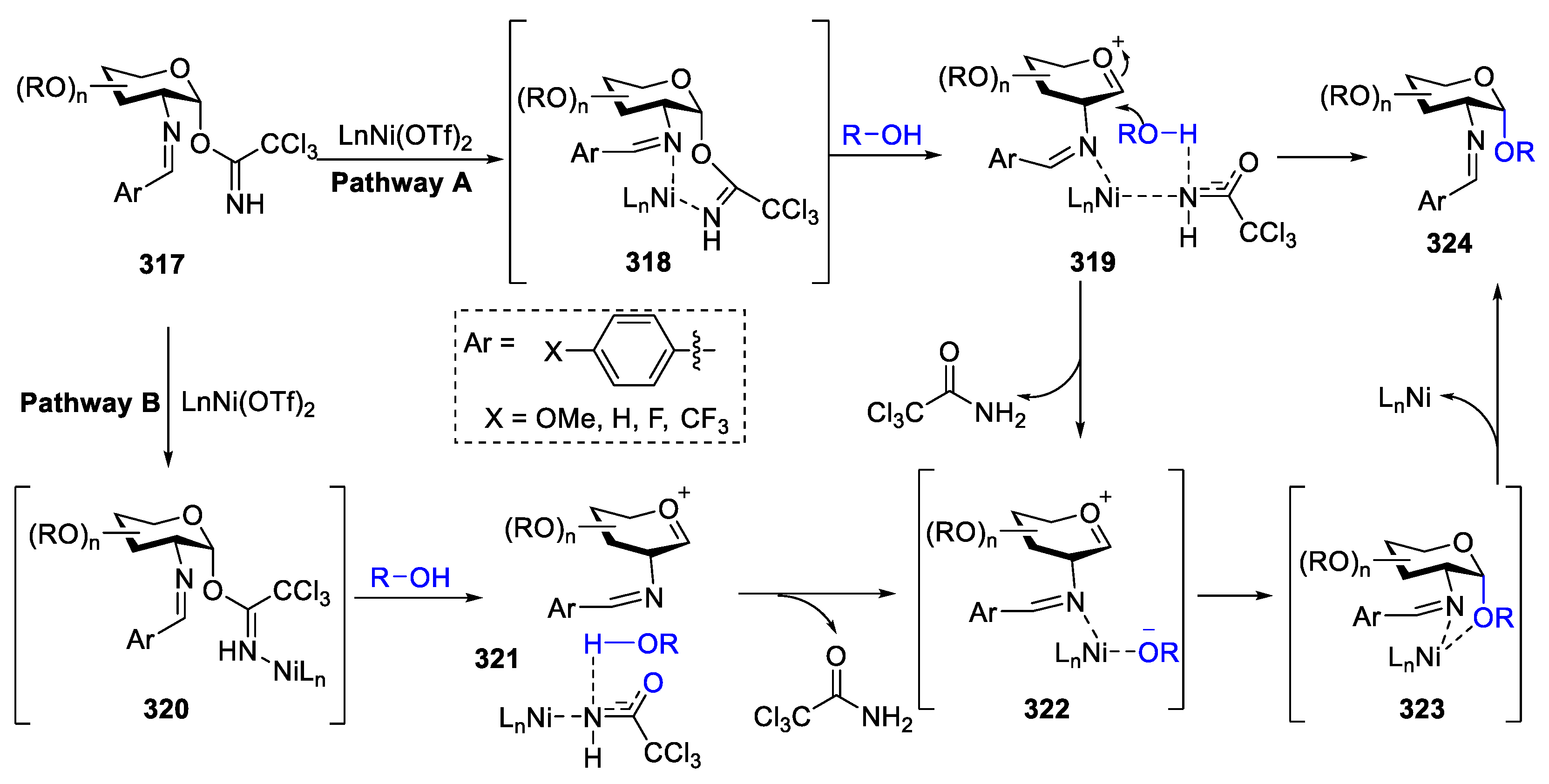
A mechanism for explaining α-selectivity of the glycosylation with cationic nickel (II) catalyst was shown in Figure 4. In pathway A, the formation of seven-member ring intermediate 318 via reversible coordination of C(2)-benzylidene nitrogen and the nitrogen of the C(1)-TCA donor 317 by nickel catalyst facilitated the 1,2-cis-glycoside. Alternatively, the cationic nickel catalyst could act as a mild Lewis acid (pathway B) to activate C(1)-TCA donor 317, promoting ionization to generate the oxocarbenium intermediate 321. Subsequently, the formation of five-membered cyclic intermediate 323 was followed by ligand exchange 322, providing the 1,2-cis-glycoside 325.
The potential of the nickel-catalyzed glycosylation strategy was further demonstrated by extending it to couple with thioglycoside acceptors. A key feature for synthesis of thioglycoside was that it was easily synthesized and generally stable under a variety of glycosylation conditions, making it compatible with other glycosylation strategies, which, in turn, facilitated it for the multistep synthesis of oligosaccharides and glycoconjugates. However, using thioglycoside as glycosyl acceptor under nickel catalyst is much more of a challenge because: (i) intermolecular aglycon transfer of the anomeric sulfide to the trichloroacetimidate donor could result in undesired thioglycoside [105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120]; and (ii) sulfur could coordinate to the cationic nickel catalyst leading to its inactivation. However, after a series of trials, the desired disaccharide products were obtained in good yields with excellent α-selectivity, and only a minor mount of undesired thioglycoside was detected. The key for the success of this transformation was using N-phenyl trifluoroacetimidate (PTFA) as donors due to their attenuated activity compared to trichloroacetimidates [121,122,123]. The scope of substrates was then expanded to the glycosylation of a variety of thioglycosides with both armed and disarmed donors [124] (Scheme 39). Three L-rhamnose acceptors equipped with different anomeric sulfides (329–331) were coupled with corresponding donors (324 and 325). Coupling of armed donor 324 with phenyl thioglycoside 141 generates disaccharide 332 in 70% yield with no undesired thioglycoside by-product was detected, albeit in lower α-selectivity (α:β = 6:1). Similarly, coupling of phenyl D-galactosyl thioglycoside 327 with both donors 325 and 326 (entries 2 and 3) provids disaccharides 333 and 334 in exclusive 55% and 61% yield with exclusive α-selectivity but a large quantity of sulfide transfer products 341 and 342 were detected (8 vs. 24%), respectively. The donor 325 then reacted with D-galactosyl thioglycoside 328. No trace amount of transfer product 341 was observed, and exclusive α-disaccharide 335 was isolated in 71% yield. The desired disaccharides 336 and 337 were obtained in 81% and 72% yield with exclusive α-selectivity, also aglycon transfer products 341 and 342 were detected in 15% and 9% yield, respectively. Compared to the acceptor 330, the coupling of bulky 2,6-dimethylphenyl (DMP) [125] acceptor 331 with donor 325 produced the desired product 340 in a slight lower yield (98% vs. 74%) with the α-only selectivity maintained but no undesired thioglycoside by-product was detected. Additionally, 2-trifluoromethylphenyl thioglycoside acceptor 330 and galactosamine acceptors 327 and 328 were also investigated, providing α-disaccharides, and aglycon transfer products were not observed. Galactosamine donors were also employed to couple with corresponding acceptors, but the desired products were obtained in lower yield and minor amounts of undesired thioglycosides were observed in some reactions.
To compare, the author also performed nickel-catalyzed glycosylation using C(2)-azido of N-phenyltrifluoroacetimidate as the glycosyl donor. It was found that the yield of disaccharide products decreased dramatically but α-only selectivity was maintained (Scheme 40). In the same case, such as with 141 as glycosyl acceptor, the major product of the reaction was thioglycoside by-product derived from the intermolecular aglycon transfer of the anomeric sulfide to N-phenyltrifluoroacetimidate.
To further explore the utility of this method, the sequential formation of trisaccharides was performed (Scheme 41). Coupling disaccharide thioglycoside 338 obtained from nickel-catalyzed glycosylation with acceptor 132 under NIS-AgOTf activation condition at 35 °C produced trisaccharide adduct 349 in 84% yield with the a slight α-selectivity (α:β = 3:2). Similarly, trisaccharide 351 was obtained by coupling disaccharide thioglycoside 350 with acceptor 132 under the same conditions. Subsequent deprotection of imine by treatment of 351 with HCl, acetone/DCM, followed by acetylation in the presence of acetic anhydride and pyridine provided corresponding product 352 in quantitative yield.
2.4.2. Synthesis of Biologically Active Glycans via Nickel-Catalyzed Glycosylation
Nguyen’s group further extended nickel-catalyzed glycosylation to synthesis of several biologically relevant glycans and glycoconjugates including mycothiol, precursors of GPI anchor and TN antigen.
Synthesis of Mycothiol
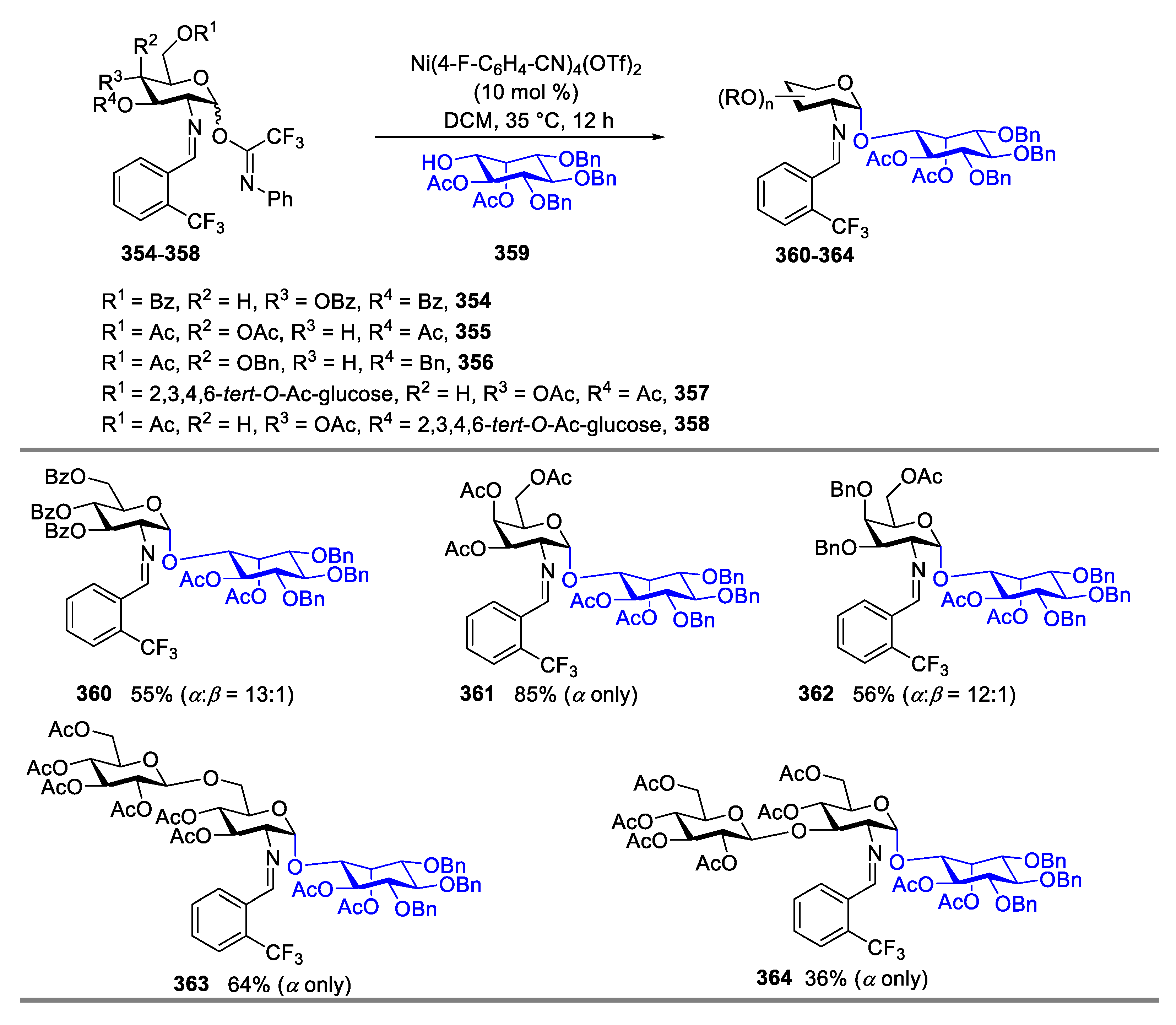
Mycothiol (MSH, 353) (Figure 5) was first isolated as a dimer (polymerized by disulfide bond) by Yasuhiro Yamada from Streptomyces sp. AJ9463 in 1994. It is a low-molecular-weight thiol composed by an unusual N-acetylcysteine amide-linked to 1-D-myo-inosityl-2-amino-2-deoxy-α-D-glucopyranoside (GlcN-α(1-1)-Ins). MSH is used for protection of actinomycetes against foreign electrophilic reagents, such as oxidants and radicals [126,127,128]. Most actinomycetes produced MSH as the major thiol, but at millimolar levels, and this limited its application in the study on the function of MSH, which, in turn, promoted the development of total synthesis of MSH. Previously, except for the intramolecular α-glucosaminidation [129] and desymmetrization of meso-inositol strategies [130], the existing chemical synthesis provided the desired pseudo-disaccharide of MSH in poor-to-moderate selectivity (α:β = 1:1 to 6:1) [129,130,131,132,133]. Accordingly, the nickel-catalyzed method was employed in the synthesis of mycothiol core pseudosaccharides via coupling a mixture of α- and β-isomers of C(2)-N-substituted benzylidenamino donors with C(1)-hydroxyl inositol acceptor 359 [134].
As shown in Scheme 42, under standard nickel-catalyzed glycosylation conditions, the mycothiol core analogs were isolated in moderate-to-good yield (36–64%) with excellent selectivity (α:β = 12:1 to α only). When the tribenzoylated substrate 354 was employed as the donor, the anomeric selectivity of the desired product 360 is α:β = 13:1. Additionally, coupling the galactosamine dibenzylated donor 356 with acceptor 359 generated the desired product 362 in 56% yield with α:β = 12:1. Even when 1,6-linked disaccharide 357 was employed as donor, the desired pseudotrisaccharide 362 was still produced in 64% yield with exclusively α-selectivity. Similarly, coupling 1,4-linked disaccharide 357 with 359 produced pseudotrisaccharide 364 in 64% yield and exclusive α-selectivity as well.
To synthesise mycothiol 353, the 2-trifluoromethyl-benzylidene in pseudodisaccharide 365 was removed in the presence of 5N HCl to obtain free ammonium salt 366. Hydrogenolysis of 366 with Pd(OH)2/C and H2 [135], which was followed by deacetylation, generated 367 in 55% yield over two steps. Coupling 367 with cysteine residue 368 formed mycothiol 353 following the previously reported procedures [129,130] (Scheme 43).
Synthesis of GPI Anchor
The GPI anchor, a glycophospholipid, is post-translationally tethered to a protein for attachment to a cell membrane [136]. These anchored proteins play a pivotal role in biochemical processes, such as signal transduction, immune response, and cancer metastasis [137,138]. All GPI structures share the same basic core, including a phosphoethanolamine linker, a tetrasaccharide attached to the C(6)-position of myo-inositol, and phospholipid tail [139]. Despite the syntheses of GPI anchors have been reported extensively, there are several challenges to stereoselectively form the 1,2-cis-glycosidic bond between the glucosamine unit and the myo-inositol component [140,141,142]. The cationic nickel (II) catalysis developed by Nguyen’s group is thought to be suitable for the GPI anchor core due to its similarity with mycothiol 353 (Scheme 44) [139]. Coupling C(4)-TES-C(2)-N-benzylidene donor 369 with C(6)-hydroxyl myo-inositol 370 under the standard condition generated the desired pseudodisaccharide 371 in 61% yield with α:β = 11:1. Subsequent removal of the TES group with TBAF generated the disaccharide 372. Finally, glycosylation of 372 with tetrabenzylated D-mannose trichloroacetimidate 373 afforded the desired GPI core pseudotrisaccharide 374 in 56% yield.
Synthesis of TN Antigen
TN antigen is a core structure in O-linked glycoproteins, which consists of the linkage of an N-acetyl-galactosamine (GalNAc) and the hydroxyl group of serine or threonine via α-glycosidic bond [143]. TN antigen 375 and some other analogs, such as TF antigen 376 and STN antigen 377 (Figure 6), are widely distributed on cell-surface mucin glycoproteins, which participate in cell-adhesion events associated with cancer metastasis, having potential as a biomarker or as a vaccine against cancer [144,145,146].
Early work employed the C(2)-oxazolidinone and the C(2)-azido as donors to couple with the acceptor, affording a mixture of anomers with the ratio of α:β ranged from 1:1 to 4:1 [78,147]. Large-scale synthesis of TN antigen core in good yield with α-selectivity could be achieved under the condition of cationic nickel (II) catalysis (Scheme 45). Two small-scale (Scheme 45a,b) and scale-up reactions (Scheme 45c) employing donor 355 were performed by using 10 mol% of Ni(4-F-C6H4-CN)4(OTf)2 as the catalyst [148]. These scale-up reactions without loss in yield and selectivity showed that the cationic nickel (II) catalyst might be suitable for formation of 1,2-cis-aminoglycoside in large-scale industry production.
Finally, gram-scale synthesis of TN antigen from 379 was performed (Scheme 46). Treatment of 379 with HCl(g) (in situ formed from the reaction between AcCl and MeOH) followed by the addition of Ac2O-generated N-acetylated 380. Full deprotection of 378 with 0.2N NaOH in methanol produced 0.4 g TN antigen 375 in 82% yield. Additionally, the synthesis of TN/TF antigens was also completed by Nguyen’s group via the nickel methodology in 2016 [149] and 2019 [150].
3. Glycosylation of 3-Amino-3-Deoxysugars and 4-Amino-4-Deoxysugars
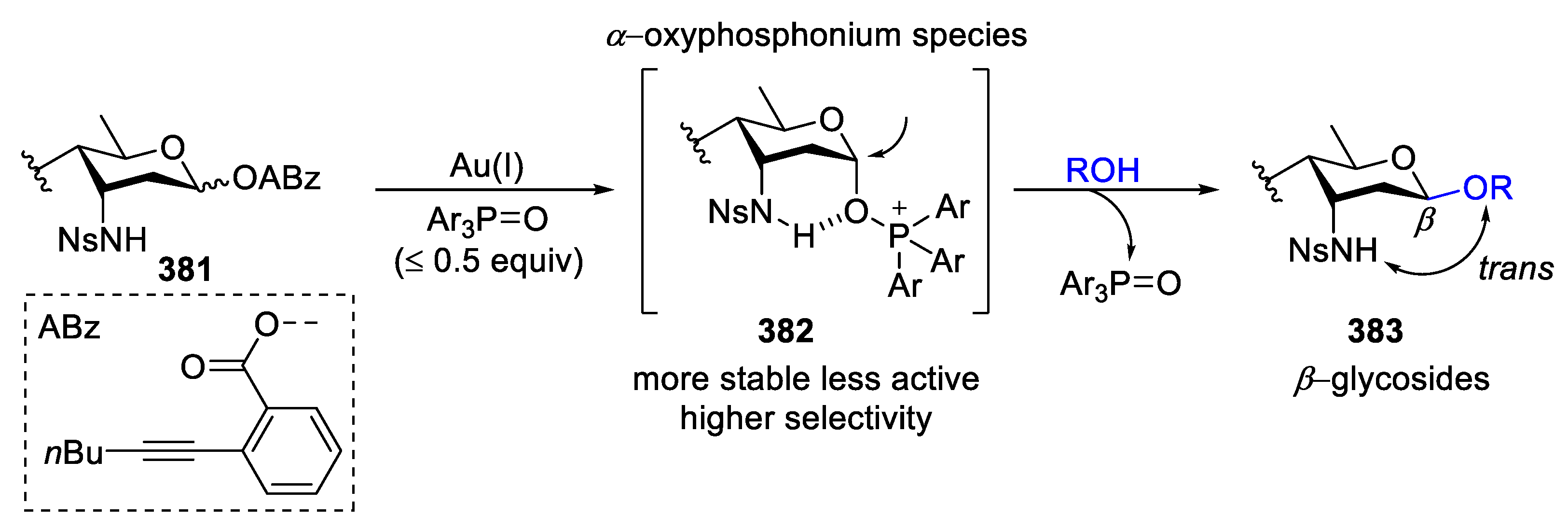
Over the past few years, Wan’s group has been working on the development of novel and effective methodologies for stereoselective formation of 3-aminoglycoside and applying the methods developed by them for the synthesis of natural products containing 3-amino-3-deoxysugars [151,152]. Inspired by their earlier report where an intramolecular hydrogen bond (HB) between a C3-nosyl group and an α-hydroxyl group on the anomeric carbon was observed in X-ray crystallography [153], they reported an HB-assisted protocol for β-selective glycosylation of rare 3-amino sugars (Scheme 47) [154]. By employment of 381 as the glycosyl donor and phosphine oxide as an exogenous nucleophile, an α-oxyphosphonium species 382 was generated, in which there was an intramolecular hydrogen bond between the C3-axial sulfonamide group and the oxygen of the α-aligned phosphine oxide. Oxyphosphonium species 382 blocked α-face leading to nucleophiles attacked to 382 from β-face, which, in turn, resulted in β-selective glycosylation.
This H-bond-assisted glycosylation reaction was found to have broad application in the construction of β-glycosidic bonds (Scheme 48). For example, coupling of donor 363a–d with bulky primary alcohol offered glycosylation products in better selectivity than that with the less sterically-hindered and electron-rich primary alcohols (366a and 366c) due to the latter one being a good H-bond acceptor to compete with the phosphine oxide additive. Secondary alcohols generally gave the products with β-selectivity. Additionally, this strategy was also extended to modification of natural products and drugs in good yield with satisfactory-to-excellent β-selectivity (391g–391i). This strategy was also suitable for the one-pot formation of trisaccharide such as 394 with Ph3PAuBAr4F as the only catalyst. The glycosylation reaction between donor 392 and acceptor 393 occurred selectively on the 6-OH of 392 with α-selectivity in the presence of Ph3PAuBAr4F catalyst. Then, 384a and 390 were added to the resulting mixture, furnishing the 394 with exclusive β-selectivity in 52% yield over two steps; impressively, the whole process used Ph3PAuBAr4F as the only catalyst for the two glycosylation reactions.
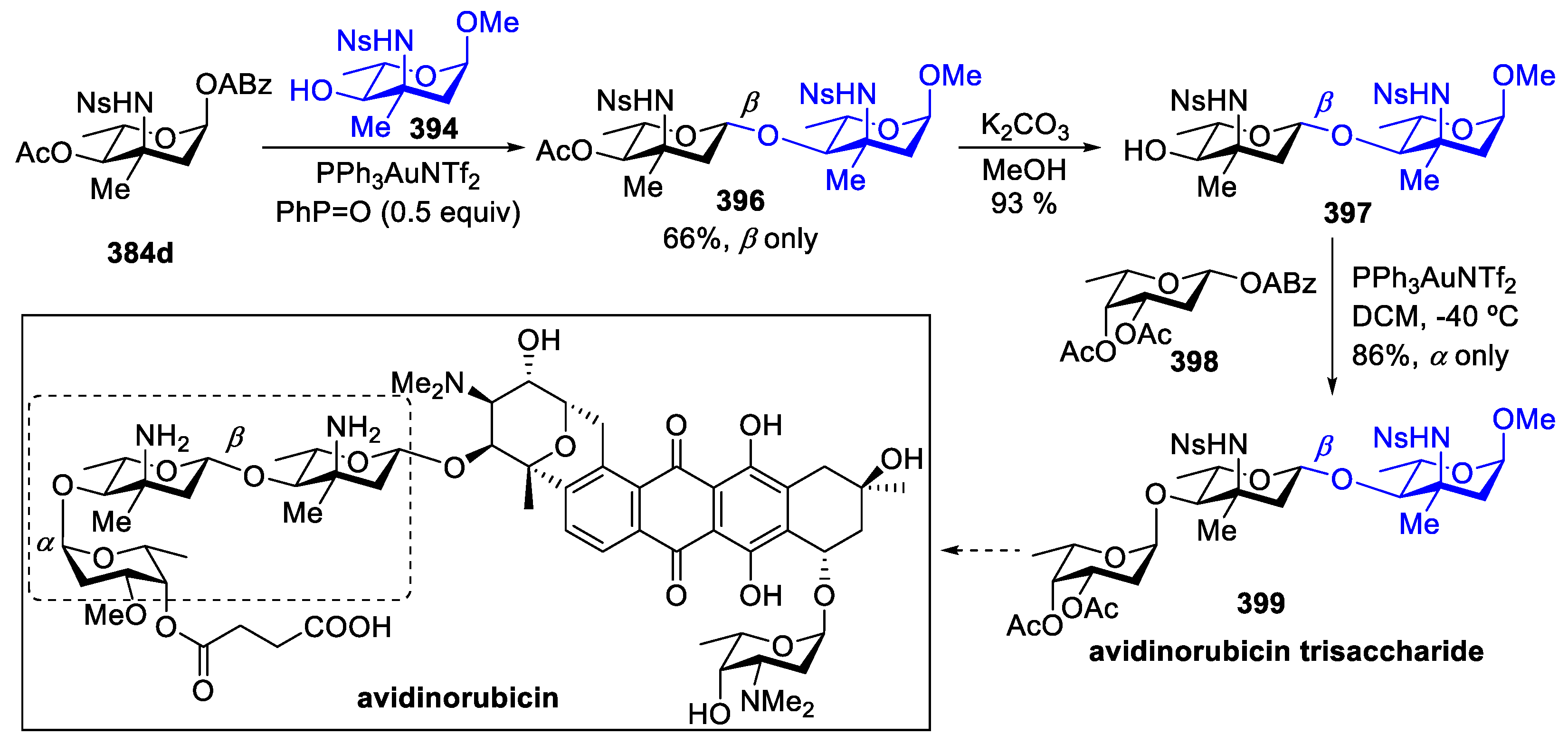
The methodology was further applied in the synthesis of trisaccharide fragment 399 of avidinorubicin. Avidinorubicin is an antibiotic isolated from the cultured broth of Streptomyces avidinii in 1991, it exhibited platelet-aggregation-inhibitory activity [155]. The synthesis of avidinorubicin is challenging due to its complex structure. Coupling 3-ADS 384d with 394 produced the desired disaccharide 396 in 66% yield with exclusive β-selectivity. Subsequent removal of the acetyl group generated 397 in 93% yield. Finally, glycosylation between 2,6-dideoxy sugar 398 and disaccharide 397 under Yu’s glycosylation conditions proceeded smoothly, generating the protected trisaccharide unit of avidinorubicin 399 in 86% yield with exclusive α-selectivity (Scheme 49).
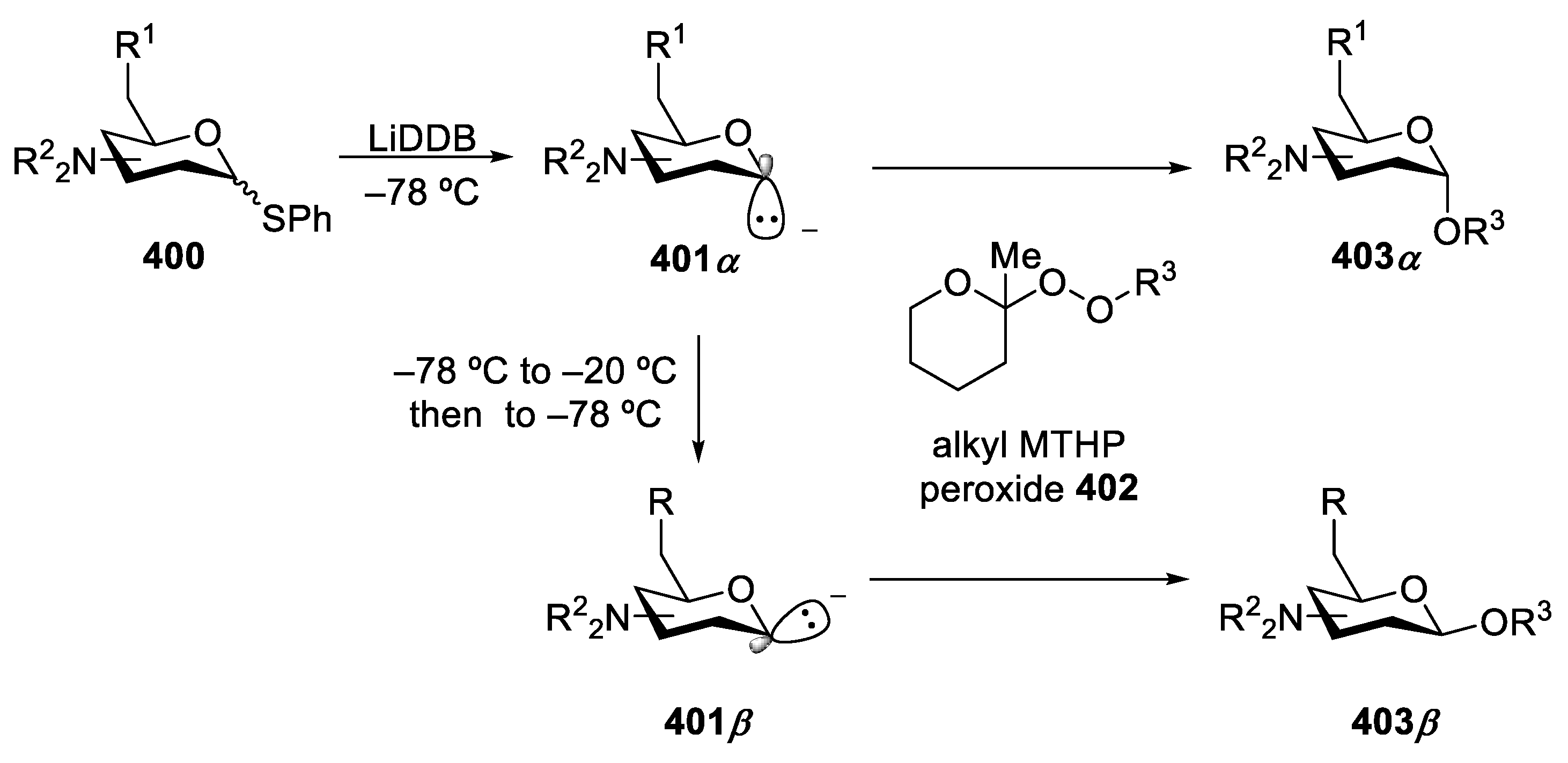
Herzon’s group reported the synthesis of deoxyaminoglycoside bearing basic amine residues in 2021 [156], based upon their early work on the synthesis of 2-deoxyglycoside by similar strategy in 2019 [157]. In this strategy, they effectively used equilibration between α-anomeric anion and β-anomeric anion by adjusting the reaction temperature. Lithium 4,4′-di-tert-butylbiphenylide (LiDBB) [158] was employed in reductive lithiation of thiophenyl glycoside 400 to form the axial (α) anion 401α as the kinetic product [159,160,161,162], then alkyl (2-methyl)-tetrahydropyranyl (MTHP) peroxide 402 [163,164] was added to the reaction mixture, resulting in alkoxenium ion transfer to generate the α-linked glycoside 403α. Warming the α anion to −20 °C followed by re-cooling to −78 °C with peroxide 404 addition formed products 403β in β-selectivity, due to the thermodynamically more stable [165] equatorial (β) anion 401β rather than the axial (α) anion 401α (Scheme 50).
As for the scope of substrates, the synthesis of α-aminoglycoside was investigated first (Scheme 51). Coupling the acosamine derivatives 404–406 with the primary MTHP electrophile 407 generates the desired disaccharides 409–411 in 79−96% yield with α-selectivity (α:β > 50:1). The secondary MTHP electrophile 406 was also employed to react with these pronucleophiles, producing the desired disaccharides 412−414 in 78−92% yield with α-selectivity (α:β > 50:1).
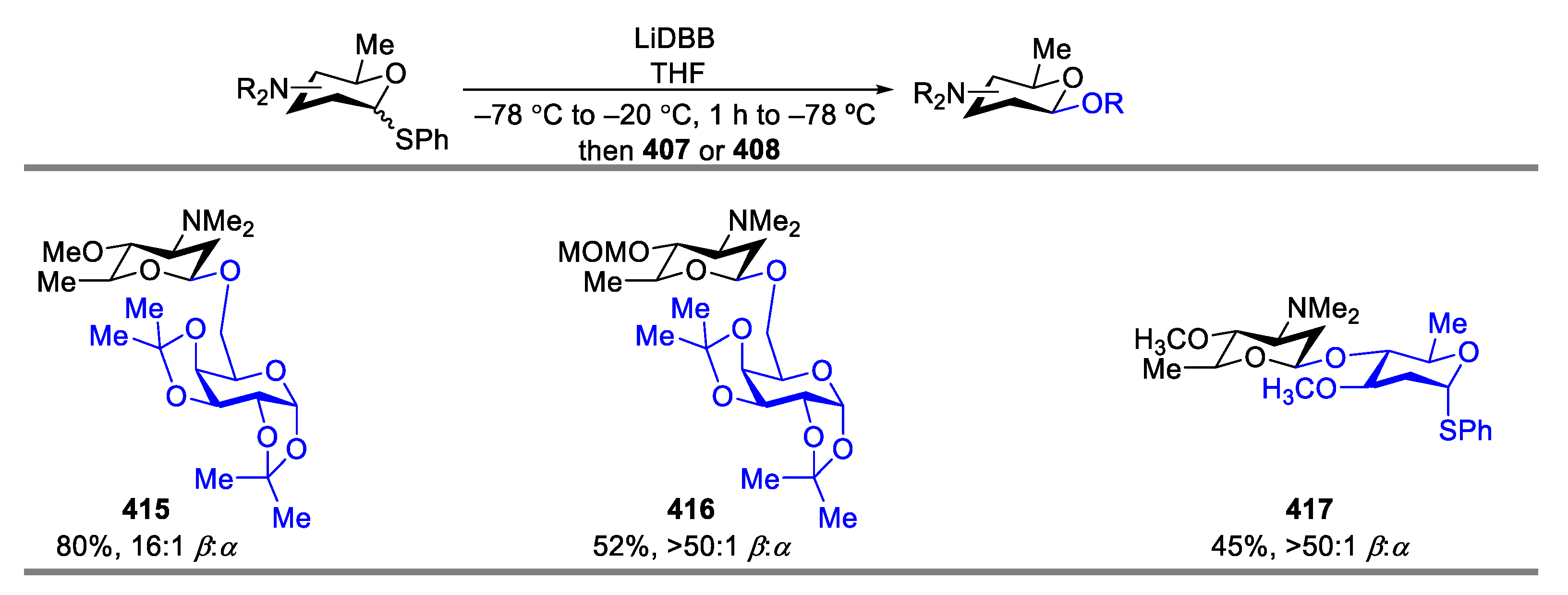
Then, the synthesis of β-aminoglycoside was investigated (Scheme 52). The disaccharide 415 was obtained in 80% yield with satisfactory β-selectivity (β:α = 16:1) using THF as the only solvent. The disaccharide 417 was obtained in 45% yield with excellent β-selectivity (β:α > 50:1). Additionally, the methoxymethyl ether derivative 405 was transformed to the disaccharide 416 in 52% yield with excellent β-selectivity (β:α > 50:1) as well.
This methodology was also suitable for glycosylation of 4-amino-4-deoxysugars [116]. Coupling of acosamine derivatives 418 and 419 with MTHP electrophile 405 and 406 under Condition A generated desired disaccharides 420–423 in 60−93% yield with α-selectivity (Scheme 53). While disaccharide 424 was obtained in 64% yield with β-selectivity (β:α > 50:1) under Condition B (see Scheme 53 for detail). Under Condition B, disaccharide 426 was obtained in 52% yield with β-selectivity (β:α > 50:1) following addition of secondary electrophile 408, and derivative 419 was transformed into disaccharide 424 in 56% yield with β-selectivity (β:α > 2.6:1).
Sialic acid (Neu5Ac) is an acidic nine carbon saccharide, which usually plays an essential role in terminating the carbohydrate portions of glycoproteins and glycolipids of mammals in the form of an α-glycoside. Silalylation via chemical methods with high α-stereoselectivity is challenging. For instance, the anomeric selectivity at the newly formed glycosidic bond is unpredictable due to the absence of a C3 participating group. On the other hand, the presence of the C1 electron-withdrawing carboxylic group could reduce the reactivity of sialic acid donor, resulting in a poor yield and formation of a significant amount of undesirable 2,3-glycal side-product. In view of the above challenges, Yang’s group developed an efficient α-sialylation methodology for various primary, secondary, and tertiary alcohol acceptors by using C4-Pico or 4-nitropicoloyl as directing group (Scheme 54) [166].
As for the scope of substrates, it was found that the combination of C4 Pico or 4-nitropicoloyl groups with the DCM/CH3CN [167,168] mixture solvent system is more effective and selective for α-glycosylation (Figure 7). Additionally, glycosylation of donor 432 with various acceptors all generated the desired products with satisfactory α-selectivity.
In order to further explore this methodology, the synthesis of sialylated trisaccharide 446 was examined (Scheme 55). Coupling donor 432 with acceptor 441 provided sialylated disaccharide 442 in 52% yield with exclusive α-selectivity. Then, removal of the two 2-naphthylmethyl (Nap) protecting groups in 442 in the presence of 2,3-dichloro-5,6-dicyanobenzo-1,4-quinone (DDQ) gave the desired disaccharide triol 443 in 91% yield, which was followed by acylation with benzoyl chloride (BzCl) to produce 444 in 85% yield. Subsequently, deprotection of 1-O-p-methoxyphenyl group in 444 with ceric ammonium nitrate (CAN) followed by imidation with trichloroacetonitrile (CCl3CN) produced trichloroacetimidate 445 in 51% over two steps as inseparable α:β (1:1) isomers. Finally, coupling trichloroacetimidate 445 with monosaccharide alcohol 446 [169] under the activation of trimethylsilyl trifluoromethanesulfonate (TMSOTf) in DCM at −78 °C, generated the target trisaccharide 447 in 51% yield with exclusive α-selectivity.
Additionally, similar strategies of glycosylation with various picoloylated sialyl donors were reported by Cristina De Meo’s group [170,171] and other strategies are also reported [172,173,174,175]. There are also several elegant reviews about glycosylation of sialic acid [176,177,178]; we will not include these herein.
4. Conclusions
Amino sugars exist widely in nature and have a range of biological activities. The fine distinctions in their glycan structures may lead to the difference of the activities and, thus, can be exploited for the development of drugs and vaccines. The new methodologies and strategies for stereoselective aminoglycoside construction and convergent oligosaccharide assembly are critical for the development of carbohydrate chemistry. Many new leaving groups and catalyst systems have been developed with the goal to develop a versatile method for stereoselective glycosylation of amino sugars.
In this review, we summarized the advances of the stereoselective glycosylation protocols of amino sugars. A number of glycosylation strategies under mild conditions with potential applications mediated or catalyzed by transition-metal and (or) organic catalysts have been developed for stereoselective glycosylation of amino sugars. These methodologies render the glycosylation reaction more effective and extend the scope of donors and acceptors with a broad range of protecting groups, being no longer only dependent on neighboring group participation or anomeric effect. However, the results of these glycosylation methods are also affected by other factors, such as solvent effect, remote participation, steric hindrance, and conformational constraints, as well. After generous traditional approaches for the development of the synthesis of aminoglycosides, glycosidation of amino sugars via glycosyl radical intermediates has been a promising method and powerful tool, owing to its inherent features, including mild reaction conditions, broad functional groups tolerance, and being free of side reactions such as elimination and/or epimerization. Notably, the stereoselective formation of 1,2-cis-aminoglycosides via radical activation of glycosyl precursors is much less developed compared to conventional ionic activation strategies. In this context, it remains highly desirable to exploit general and user-friendly photocatalyzed or electrocatalyzed glycosylation methods to construct the challenging 1,2-cis-aminoglycosidic bonds that avoid sensitive reagents, strong (Lewis) acids, precious (metal) catalysts, or labile substrates.
Author Contributions
Conceptualization X.M.; Original draft was prepared by J.Y.; Review and editing was done by X.M., J.Y. and D.X. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful for the financial support from the National Natural Science Foundation of China (22001246), the Biological Resources Program (KFJ-BRP-008) at the Chinese Academy of Sciences, and the Sichuan Science and Technology Program (2022ZYD0047).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Ac | acetyl |
| All | allyl |
| Ar | ary (substituted aromatic ring) |
| ABz | o-hexynylbenzoic acid |
| Bn | benzyl |
| Boc | t-butoxycarbonyl |
| BSM | benzenesulfinyl morpholine |
| BSP | 1-benzensulfinylpiperidine |
| Bz | benzoyl |
| nBu | n-butyl |
| tBu | t-butyl |
| Cbz | benzyloxycarbonyl |
| DCM | dichloromethane |
| DNs | 2,4-dinitrobenzenesulfonyl |
| Et | ethyl |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| HFIP | 1,1,1,3,3,3-hexafluoro-2-propanol |
| KHMDS | potassium bis(trimethylsilyl)amide |
| Me | methyl |
| NIS | N-iodosuccinimide |
| Ns | p-nitrobenzene sulfonyl |
| Phth | phthaloyl |
| PMB (MP) | p-methoxybenzyl |
| iPr | isopropyl |
| Ser | L-serine |
| TBAF | tetrabutylammonium fluoride |
| TBS | t-butyldimethylsilyl |
| TES | triethylsilane |
| Tf | trifluoromethanesulfony |
| Thr | L-threonine |
| TIPS | triisopropylsilyl |
References
- Gannett, C.; Banks, P.; Chuong, C.; Weger-Lucarelli, J.; Mevers, E.; Lowell, A.N. Semisynthetic Blasticidin S Ester Derivatives Show Enhanced Antibiotic Activity. RSC Med. Chem. 2023, 14, 782–789. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Zhou, T.; Guo, Y.; Zhao, X.; Wu, Y. Molecular Regulation of Host Defense Responses Mediated by Biological Anti-TMV Agent Ningnanmycin. Viruses 2019, 11, 815. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.-H.; Luo, D.; Shu, D.; Zhong, J.; Tan, H. Development of an Intergeneric Conjugal Transfer System for Xinaomycins-Producing Streptomyces noursei Xinao-4. Int. J. Mol. Sci. 2014, 15, 12217–12230. [Google Scholar] [CrossRef] [PubMed]
- Flatt, P.M.; Mahmud, T. Biosynthesis of Aminocyclitol-Aminoglycoside Antibiotics and Related Compounds. Nat. Prod. Rep. 2007, 24, 358–392. [Google Scholar] [CrossRef] [PubMed]
- Behera, A.; Kulkarni, S. Chemical Synthesis of Rare, Deoxy-Amino Sugars Containing Bacterial Glycoconjugates as Potential Vaccine Candidates. Molecules 2018, 23, 1997. [Google Scholar] [CrossRef]
- Bennett, C.S.; Galan, M.C. Methods for 2-Deoxyglycoside Synthesis. Chem. Rev. 2018, 118, 7931–7985. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.C.; Seitz, S.P.; Pavia, M.R. Carbohydrates in Organic Synthesis. Synthesis of 16-Membered-Ring Macrolide Antibiotics. 6. Total Synthesis of O-Mycinosyltylonolide: Coupling of Key Intermediates and Macrocyclization. J. Am. Chem. Soc. 1982, 104, 2030–2031. [Google Scholar] [CrossRef]
- Suzuki, K.; Maeta, H.; Matsumoto, T.; Tsuchihashi, L.G.-I. New Glycosidation Reaction 2. Preparation of 1-Fluoro-D-Desosamine Derivative and Its Efficient Glycosidation by the Use of Cp2HfCl2-AgClO4 as the Activator. Tetrahedron Lett. 1988, 29, 3571–3574. [Google Scholar] [CrossRef]
- Bai, Y.; Shen, X.; Li, Y.; Dai, M. Total Synthesis of (-)-Spinosyn A via Carbonylative Macrolactonization. J. Am. Chem. Soc. 2016, 138, 10838–10841. [Google Scholar] [CrossRef] [Green Version]
- Balthaser, B.R.; McDonald, F.E. Brønsted Acid-Promoted Glycosylations of Disaccharide Glycal Substructures of the Saccharomicins. Org. Lett. 2009, 11, 4850–4853. [Google Scholar] [CrossRef] [Green Version]
- Bylsma, M.; Bennett, C.S. Stereospecific Synthesis of the Saccharosamine-Rhamnose-Fucose Fragment Present in Saccharomicin B. Org. Lett. 2018, 20, 4695–4698. [Google Scholar] [CrossRef]
- Skarbek, K.; Milewska, M.J. Biosynthetic and Synthetic Access to Amino Sugars. Carbohydr. Res. 2016, 434, 44–71. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, H.; Hua, M.; Jiao, Y.; He, H.; Liu, M.; Huang, N.; Zou, K. Direct N-Glycosylation of Amides/Amines with Glycal Donors. J. Org. Chem. 2020, 85, 7485–7493. [Google Scholar] [CrossRef]
- Sangwan, R.; Khanam, A.; Mandal, P.K. An Overview on the Chemical N-Functionalization of Sugars and Formation of N-Glycosides. Eur. J. Org. Chem. 2020, 2020, 5949–5977. [Google Scholar] [CrossRef]
- Manabe, S. Chapter Twenty—The Synthesis of 1,2-cis-Amino Containing Oligosaccharides toward Biological Investigation. In Methods in Enzymology; Fukuda, M., Ed.; Academic Press: Cambridge, MA, USA, 2010; pp. 413–435. [Google Scholar]
- Bongat, A.F.G.; Demchenko, A.V. Recent Trends in the Synthesis of O-glycoside of 2-Amino-2-Deoxysugars. Carbohydr. Res. 2007, 342, 374–406. [Google Scholar] [CrossRef]
- Petitou, M.; van Boeckel, C.A.A. A Synthetic Antithrombin III Binding Pentasaccharide Is Now a Drug! What Comes Next? Angew. Chem. Int. Ed. 2004, 43, 3118–3133. [Google Scholar] [CrossRef]
- Hakomori, S.-I. Tumor-Associated Carbohydrate Antigens. Amnu. Rev. Immunol. 1984, 2, 103–126. [Google Scholar] [CrossRef]
- Danishefsky, S.J.; Allen, J.R. From the Laboratory to the Clinic: A Retrospective on Fully Synthetic Carbohydrate-Based Anticancer Vaccines. Angew. Chem. Int. Ed. 2000, 39, 836–863. [Google Scholar] [CrossRef]
- Dwek, R.A. Glycobiology: Toward Understanding the Function of Sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef]
- Werz, D.B.; Ranzinger, R.; Herget, S.; Adibekian, A.; von der Lieth, C.-W.; Seeberger, P.H. Exploring the Structural Diversity of Mammalian Carbohydrates (“Glycospace”) by Statistical Databank Analysis. ACS Chem. Biol. 2007, 2, 685–691. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked β-N-Acetylglucosamine on Nucleocytoplasmic Proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Banoub, J.; Boullanger, P.; Lafont, D. Synthesis of Oligosaccharides of 2-Amino-2-Deoxy Sugars. Chem. Rev. 1992, 92, 1167–1195. [Google Scholar] [CrossRef]
- Kerns, R.J.; Wei, P. Synthetic Methods to Incorporate α-Linked 2-Amino-2-Deoxy-D-Glucopyranoside and 2-Amino-2-Deoxy-D-Galactopyranoside Residues into Glycoconjugate Structures. In Glycobiology and Drug Design; American Chemical Society: Washington, DC, USA, 2012; pp. 235–263. [Google Scholar]
- Beau, J.M.; Boyer, F.D.; Norsikian, S.; Urban, D.; Vauzeilles, B.; Xolin, A. Glycosylation: The Direct Synthesis of 2-Acetamido-2-Deoxy-Sugar glycoside. Eur. J. Org. Chem. 2018, 2018, 5795–5814. [Google Scholar] [CrossRef]
- Wang, X.; Wang, P.; Li, D.; Li, M. 2,4-Dinitrobenzenesulfonamide-Directed SN2-Type Displacement Reaction Enables Synthesis of β-D-Glycosaminosides. Org. Lett. 2019, 21, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Ding, H.; Peng, L.-C.; Fang, X.-Y.; Qin, Y.-Y.; Mu, Q.-Q.; Liu, X.-W. Sweet Strain Release: Donor–Acceptor Cyclopropane Mediated Glycosylation. CCS Chem. 2023; in press. [Google Scholar] [CrossRef]
- Orgueira, H.A.; Bartolozzi, A.; Schell, P.; Litjens, R.; Palmacci, E.R.; Seeberger, P.H. Modular Synthesis of Heparin Oligosaccharides. Chem. Eur. J. 2003, 9, 140–169. [Google Scholar] [CrossRef]
- Tingoli, M.; Tiecco, M.; Chianelli, D.; Balducci, R.; Temperini, A. Novel Azido-Phenylselenenylation of Double Bonds. Evidence for a free-radical process. J. Org. Chem. 1991, 56, 809–6813. [Google Scholar] [CrossRef]
- Koto, S.; Asami, K.; Hirooka, M.; Nagura, K.; Takizawa, M.; Yamamoto, S.; Okamoto, N.; Sato, M.; Tajima, H.; Yoshida, T.; et al. Glycosylation using 2-Azido-3,4,6-Tri-O-Benzyl-2-Deoxy-D-Glucose, -Galactose, and -Mannose with the Aid of p-Nitrobenzenesulfonyl Chloride Silver Trifluoromethanesulfonate Triethylamine System. Bull. Chem. Soc. Jpn. 1999, 72, 765–777. [Google Scholar] [CrossRef]
- Singh, Y.; Wang, T.H.; Demchenko, A.V. Direct Glycosidation of 2-Azido-2-Deoxyglycosyl Nitrates. Eur. J. Org. Chem. 2019, 2019, 6413–6416. [Google Scholar] [CrossRef]
- Jeanneret, R.A.; Johnson, S.E.; Galan, M.C. Conformationally Constrained Glycosyl Donors as Tools to Control Glycosylation Outcomes. J. Org. Chem. 2020, 85, 15801–15826. [Google Scholar] [CrossRef]
- Bousquet, E.; Khitri, M.; Lay, L.; Nicotra, F.; Panza, L.; Russo, G. Capsular Polysaccharide of Streptococcus Pneumoniae Type 19F: Synthesis of the Repeating Unit. Carbohydr. Res. 1998, 311, 171–181. [Google Scholar] [CrossRef]
- Crich, D.; Cai, W. Chemistry of 4,6-O-Benzylidene-D-glycopyranosyl Triflates: Contrasting Behavior between the Gluco and Manno Series. J. Org. Chem. 1999, 64, 4926–4930. [Google Scholar] [CrossRef]
- van der Vorm, S.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. Stereoselectivity of Conformationally Restricted Glucosazide Donors. J. Org. Chem. 2017, 82, 4793–4811. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Guo, Z. Convergent Synthesis of An Inner Core GPI of Sperm CD52. Bioorg. Med. Chem. Lett. 2002, 12, 2015–2018. [Google Scholar] [CrossRef]
- Plattner, C.; Höfener, M.; Sewald, N. One-Pot Azidochlorination of Glycals. Org. Lett. 2011, 13, 545–547. [Google Scholar] [CrossRef]
- Lemieux, R.U.; Ratcliffe, R.M. The Azidonitration of Tri-O-Acetyl-D-Galactal. Can. J. Chem. 1979, 57, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Broddefalk, J.; Nilsson, U.; Kihlberg, J. An Improved Synthesis of 3,4,6-Tri-O-Acetyl-2-Azido-2-Deoxy-α-D-Galactopyranosyl Bromide: A Key Component for Synthesis of Glycopeptides and Glycolipids. J. Carbohydr. Chem. 1994, 13, 129–132. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin–Protein Interactions. Angew. Chem. Int. Ed. 2002, 41, 390–412. [Google Scholar] [CrossRef]
- Varki, A. Biological Roles of Oligosaccharides: All of the Theories are Correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Orgueira, H.A.; Bartolozzi, A.; Schell, P.; Seeberger, P.H. Conformational Locking of the Glycosyl Acceptor for Stereocontrol in the Key Step in the Synthesis of Heparin. Angew. Chem. Int. Ed. 2002, 41, 2128–2131. [Google Scholar] [CrossRef]
- Park, J.; Kawatkar, S.; Kim, J.-H.; Boons, G.-J. Stereoselective Glycosylations of 2-Azido-2-Deoxy-Glucosides using Intermediate Sulfonium Ions. Org. Lett. 2007, 9, 1959–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kärkkäinen, T.S.; Ravindranathan Kartha, K.P.; MacMillan, D.; Field, R.A. Iodine-Mediated Glycosylation En Route to Mucin-Related Glyco-Aminoacids and Glycopeptides. Carbohydr. Res. 2008, 343, 1830–1834. [Google Scholar] [CrossRef] [PubMed]
- Kurfiřt, M.; Lucie, Č.Š.A.; Cuřínová, P.; Hamala, V.; Karban, J. Development of α-Selective Glycosylation for the Synthesis of Deoxyfluorinated TN Antigen Analogues. J. Org. Chem. 2021, 86, 5073–5090. [Google Scholar] [CrossRef] [PubMed]
- Li, Z. Computational Study of the Influence of Cyclic Protecting Groups in Stereoselectivity of Glycosylation Reactions. Carbohydr. Res. 2010, 345, 1952–1957. [Google Scholar] [CrossRef] [PubMed]
- Kalikanda, J.; Li, Z. Study of the Stereoselectivity of 2-Azido-2-Deoxygalactosyl Donors: Remote Protecting Group Effects and Temperature Dependency. J. Org. Chem. 2011, 76, 5207–5218. [Google Scholar] [CrossRef]
- Ngoje, G.; Li, Z. Study of the Stereoselectivity of 2-Azido-2-Deoxyglucosyl Donors: Protecting Group Effects. Org. Biomol. Chem. 2013, 11, 1879–1886. [Google Scholar] [CrossRef]
- Ngoje, G.; Addae, J.; Kaur, H.; Li, Z. Development of Highly Stereoselective GalN3 Donors and Their Application in the Chemical Synthesis of Precursors of Tn Antigen. Org. Biomol. Chem. 2011, 9, 6825–6831. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. Reagent Controlled Glycosylations for the Assembly of Well-Defined Pel Oligosaccharides. J. Org. Chem. 2020, 85, 15872–15884. [Google Scholar] [CrossRef]
- Lemieux, R.U.; Nagabhushan, T.L.; O’Neill, I.K. The Reactions of Nitrosyl Chloride and Dinitrogen Tetroxide with Acetylated Glycals. Acetylated 2-Deoxy-2-Nitroso-α-D-Hexopyranosyl Chlorides and Titrates and Acetylated 2-Nitroglycals. Can. J. Chem. 1968, 46, 413–418. [Google Scholar] [CrossRef]
- Khodair, A.I.; Winterfeld, G.A.; Schmidt, R.R. Conjugate Addition of Phenols to 2-Nitrogalactal—Synthesis of O-(2-Acetamido-2-Deoxygalactosyl)Tyrosine. Eur. J. Org. Chem. 2003, 2003, 1847–1852. [Google Scholar] [CrossRef]
- Das, J.; Schmidt, R.R. Convenient Glycoside Synthesis of Amino Sugars: Michael-Type Addition to 2-Nnitro-D-Galactal. Eur. J. Org. Chem. 1998, 1998, 1609–1613. [Google Scholar] [CrossRef]
- Winterfeld, G.A.; Ito, Y.; Ogawa, T.; Schmidt, R.R. A Novel and Efficient Route towards α-GalNAc-Ser and α-GalNAc-Thr Building Blocks for Glycopeptide Synthesis. Eur. J. Org. Chem. 1999, 1999, 1167–1171. [Google Scholar] [CrossRef]
- Winterfeld, G.A.; Schmidt, R.R. Nitroglycal Concatenation: A Broadly Applicable and Efficient Approach to the Synthesis of Complex O-Glycans. Angew. Chem. Int. Ed. 2001, 40, 2654–2657. [Google Scholar] [CrossRef]
- Winterfeld, G.A.; Khodair, A.I.; Schmidt, R.R. O-glycosyl Amino Acids by 2-Nitrogalactal Concatenation—Synthesis of a Mucin-Type O-Glycan. Eur. J. Org. Chem. 2003, 2003, 1009–1021. [Google Scholar] [CrossRef]
- Winterfeld, G.A.; Das, J.; Schmidt, R.R. Convenient Synthesis of Nucleosides of 2-Deoxy-2-Nitro-D-Galactose and N-Acetyl-D-Galactosamine. Eur. J. Org. Chem. 2000, 2000, 3047–3050. [Google Scholar] [CrossRef]
- Shigeno, M.; Hayashi, K.; Nozawa-Kumada, K.; Kondo, Y. Phosphazene Base t-Bu-P4 Catalyzed Methoxy–Alkoxy Exchange Reaction on (Hetero)Arenes. Chem. Eur. J. 2019, 25, 6077–6081. [Google Scholar] [CrossRef]
- Shigeno, M.; Nakamura, R.; Hayashi, K.; Nozawa-Kumada, K.; Kondo, Y. Catalytic Amination of β-(Hetero)Arylethyl Ethers by Phosphazene Base t-Bu-P4. Org. Lett. 2019, 21, 6695–6699. [Google Scholar] [CrossRef]
- Pal, K.B.; Guo, A.; Das, M.; Báti, G.; Liu, X.-W. Superbase-Catalyzed Stereo- and Regioselective Glycosylation with 2-Nitroglycals: Facile Access to 2-Amino-2-deoxy-O-glycoside. ACS Catal. 2020, 10, 6707–6715. [Google Scholar] [CrossRef]
- Medina, S.; Harper, M.J.; Balmond, E.I.; Miranda, S.; Crisenza, G.E.M.; Coe, D.M.; McGarrigle, E.M.; Galan, M.C. Stereoselective Glycosylation of 2-Nitrogalactals Catalyzed by a Bifunctional Organocatalyst. Org. Lett. 2016, 18, 4222–4225. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-L.; Zhang, Y.-T.; Liu, H.-F.; Zhou, L.; Chen, J. N-Heterocyclic Carbene Catalyzed Stereoselective Glycosylation of 2-Nitrogalactals. Org. Lett. 2017, 19, 5272–5275. [Google Scholar] [CrossRef]
- Yoshida, K.; Kanoko, Y.; Takao, K. Kinetically Controlled α-Selective O-Glycosylation of Phenol Derivatives Using 2-Nitroglycals by a Bifunctional Chiral Thiourea Catalyst. Asian. J. Org. Chem. 2016, 5, 1230–1236. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, X.; Xue, Y.; Lin, X.-E.; Wang, L.; Sun, J.-S.; Zhang, Q. Stereoselective Glycosylation with Conformation-Constrained 2-Nitroglycals as Donors and Bifunctional Thiourea as Catalyst. J. Carbohydr. Chem. 2021, 40, 535–557. [Google Scholar] [CrossRef]
- Enders, D.; Balensiefer, T. Nucleophilic Carbenes in Asymmetric Organocatalysis. Acc. Chem. Res. 2004, 37, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Bindu, S.; Sreekumar, V. N-Heterocyclic Carbenes: Reagents, Not Just Ligands! Angew. Chem. Int. Ed. 2004, 43, 5130–5135. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-Heterocyclic Carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef]
- Biju, A.T.; Kuhl, N.; Glorius, F. Extending NHC-Catalysis: Coupling Aldehydes with Unconventional Reaction Partners. Acc. Chem. Res. 2011, 44, 1182–1195. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Peckelsen, K.; Thomulka, T.; Martens, J.; Berden, G.; Oomens, J.; Neudörfl, J.-M.; Breugst, M.; Meijer, A.J.H.M.; Schäfer, M.; et al. Breslow Intermediates (Amino Enols) and Their Keto Tautomers: First Gas-Phase Characterization by IR Ion Spectroscopy. Chem.Eur. J. 2021, 27, 2662–2669. [Google Scholar] [CrossRef]
- Movassaghi, M.; Schmidt, M.A. N-Heterocyclic Carbene-Catalyzed Amidation of Unactivated Esters with Amino Alcohols. Org. Lett. 2005, 7, 2453–2456. [Google Scholar] [CrossRef]
- Phillips, E.M.; Riedrich, M.; Scheidt, K.A. N-Heterocyclic Carbene-Catalyzed Conjugate Additions of Alcohols. J. Am. Chem. Soc. 2010, 132, 13179–13181. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Guo, H.; Li, Y.-Z.; Du, G.-F.; Dai, B. N-Heterocyclic Carbene-Catalyzed Formal Cross-Coupling Reaction of α-Haloenals with Thiols: Organocatalytic Construction of sp2 Carbon–Sulfur Bonds. Chem. Commun. 2014, 50, 3719–3721. [Google Scholar] [CrossRef]
- Chen, J.; Meng, S.; Wang, L.; Tang, H.; Huang, Y. Highly Enantioselective Sulfa-Michael Addition Reactions using N-Heterocyclic Carbene as A Non-Covalent Organocatalyst. Chem. Sci. 2015, 6, 4184–4189. [Google Scholar] [CrossRef] [Green Version]
- Nie, Q.; Deng, L.; Tu, Y.; Liu, H.; Sun, J.-S.; Wang, L.; Zhang, Q. Stereoselective Synthesis of 1,1′-2-Amino Thiodisaccharides by Organocatalysis. Eur. J. Org. Chem. 2022, 2022, e202201019. [Google Scholar] [CrossRef]
- Wan, Y.; Zhou, M.; Wang, L.; Hu, K.; Liu, D.; Liu, H.; Sun, J.-S.; Codée, J.D.C.; Zhang, Q. Regio- and Stereoselective Organocatalyzed Relay Glycosylations to Synthesize 2-Amino-2-deoxy-1,3-dithioglycosides. Org. Lett. 2023, 25, 3611–3617. [Google Scholar] [CrossRef]
- Benakli, K.; Zha, C.; Kerns, R.J. Oxazolidinone Protected 2-Amino-2-Deoxy-D-Glucose Derivatives as Versatile Intermediates in Stereoselective Oligosaccharide Synthesis and the Formation of α-Linked glycoside. J. Am. Chem. Soc. 2001, 123, 9461–9462. [Google Scholar] [CrossRef]
- Kerns, R.J.; Zha, C.; Benakli, K.; Liang, Y.-Z. Extended Applications and Potential Limitations of Ring-Fused 2,3-Oxazolidinone Thioglycoside in Glycoconjugate Synthesis. Tetrahedron Lett. 2003, 44, 8069–8072. [Google Scholar] [CrossRef]
- Andreotti, A.H.; Kahne, D. The effects of Glycosylation on Peptide Backbone Conformation. J. Am. Chem. Soc. 1993, 115, 3352–3353. [Google Scholar] [CrossRef]
- Jiaang, W.-T.; Chang, M.-Y.; Tseng, P.-H.; Chen, S.-T. A Concise Synthesis of the O-Glycosylated Amino acid Building Block; Using Phenyl Selenoglycoside as A Glycosyl Donor. Tetrahedron Lett. 2000, 41, 3127–3130. [Google Scholar] [CrossRef]
- Miyajima, K.; Nekado, T.; Ikeda, K.; Achiwa, K. Synthesis of Tn, Sialyl Tn and HIV-1-Derived Peptide Antigen Conjugates Having a Lipid A Analog as an Immunoadjuvant for Synthetic VAaccines. Chem. Pharm. Bull. 1998, 46, 1676–1682. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Kerns, R.J. Factors Affecting Stereocontrol during Glycosidation of 2,3-Oxazolidinone-Protected 1-Tolylthio-N-Acetyl-D-Glucosamine. J. Org. Chem. 2005, 70, 4195–4198. [Google Scholar] [CrossRef]
- Boysen, M.; Gemma, E.; Lahmann, M.; Oscarson, S. Ethyl 2-Acetamido-4,6-Di-O-Benzyl-2,3-N,O-Carbonyl-2-Deoxy-1-Thio-β-D-Glycopyranoside as a Versatile GlcNAc Donor. Chem. Commun. 2005, 92, 3044–3046. [Google Scholar] [CrossRef] [PubMed]
- Olsson, J.D.M.; Eriksson, L.; Lahmann, M.; Oscarson, S. Investigations of Glycosylation Reactions with 2-N-Acetyl-2N,3O-Oxazolidinone-Protected Glucosamine donors. J. Org. Chem. 2008, 73, 7181–7188. [Google Scholar] [CrossRef] [PubMed]
- Manabe, S.; Ishii, K.; Ito, Y. N-benzyl-2,3-Oxazolidinone as A Glycosyl Donor for Selective α-Glycosylation and One-Pot Oligosaccharide Synthesis Involving 1,2-cis-Glycosylation. J. Am. Chem. Soc. 2006, 128, 10666–10667. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Zhang, L.-H.; Ye, X.-S. Pre-activation Protocol Leading to Highly Stereoselectivity-Controllable Glycosylations of Oxazolidinone Protected Glucosamines. Chem. Commun. 2008, 5, 597–599. [Google Scholar] [CrossRef]
- Crich, D.; Smith, M.; Yao, Q.; Picione, J. 2,4,6-Tri-tert-butylpyrimidine (TTBP): A Cost Effective, Readily Available Alternative to the Hindered Base 2,6-Di-Tert-Butylpyridine and its 4-Substituted Derivatives in Glycosylation and Other Reactions. Synthesis 2001, 2001, 0323–0326. [Google Scholar] [CrossRef]
- Geng, Y.; Zhang, L.-H.; Ye, X.-S. Stereoselectivity Investigation on Glycosylation of Oxazolidinone Protected 2-Amino-2-Deoxy-D-Glucose Donors Based on Pre-Activation Protocol. Tetrahedron 2008, 64, 4949–4958. [Google Scholar] [CrossRef]
- Mensah, E.A.; Nguyen, H.M. Nickel-Catalyzed Stereoselective Formation of α-2-Deoxy-2-Amino glycoside. J. Am. Chem. Soc. 2009, 131, 8778–8780. [Google Scholar] [CrossRef]
- Mensah, E.A.; Yu, F.; Nguyen, H.M. Nickel-Catalyzed Stereoselective Glycosylation with C(2)-N-Substituted Benzylidene D-Glucosamine and Galactosamine Trichloroacetimidates for the Formation of 1,2-cis-2-Amino glycoside. Applications to the Synthesis of Heparin Disaccharides, GPI Anchor Pseudodisaccharides, and α-GalNAc. J. Am. Chem. Soc. 2010, 132, 14288–14302. [Google Scholar]
- Mensah, E.A.; Azzarelli, J.M.; Nguyen, H.M. Palladium-Controlled β-Selective Glycosylation in the Absence of the C(2)-Ester Participatory Group. J. Org. Chem. 2009, 74, 1650–1657. [Google Scholar]
- Marra, A.; Sinaÿ, P. N-p-methoxybenzylidene Derivatives of 2-Amino-2-Deoxy-D-Glucose as Glycosyl Donors: A Reinvestigation. Carbohydr. Res. 1990, 200, 319–337. [Google Scholar] [CrossRef]
- van Boeckel, C.A.A.; Petitou, M. The Unique Antithrombin III Binding Domain of Heparin: A Lead to New Synthetic Antithrombotics. Angew. Chem. Int. Ed. 1993, 32, 1671–1690. [Google Scholar] [CrossRef]
- Arungundram, S.; Al-Mafraji, K.; Asong, J.; Leach, F.E.; Amster, I.J.; Venot, A.; Turnbull, J.E.; Boons, G.-J. Modular Synthesis of Heparan Sulfate Oligosaccharides for Structure−Activity Relationship Studies. J. Am. Chem. Soc. 2009, 131, 17394–17405. [Google Scholar] [CrossRef] [Green Version]
- Paulick, M.G.; Bertozzi, C.R. The Glycosylphosphatidylinositol Anchor: A Complex Membrane-Anchoring Structure for Proteins. Biochemistry 2008, 47, 6991–7000. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, M.A. The Structure, Biosynthesis and Functions of Glycosylphosphatidylinositol Anchors, and the Contributions of Trypanosome Research. J. Cell Sci. 1999, 112, 2799–2809. [Google Scholar] [CrossRef]
- Tiede, A.; Bastisch, I.; Schubert, J.; Orlean, P.; Schmidt, R.E. Biosynthesis of Glycosylphosphatidylinositols in Mammals and Unicellular Microbes. Biol. Chem. 1999, 380, 503–524. [Google Scholar] [CrossRef]
- Nosjean, O.; Briolay, A.; Roux, B. Mammalian GPI Proteins: Sorting, Membrane Residence and Functions. Biochim. Biophys. Acta 1997, 1331, 153–186. [Google Scholar] [CrossRef]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Tang, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless Prion Protein Pesults in Infectious Amyloid Disease without Clinical Scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Bishop, L. Chemical synthesis of GPIs and GPI-Anchored Glycopeptides. Eur. J. Org. Chem. 2004, 2004, 3585–3596. [Google Scholar] [CrossRef]
- Udodong, U.E.; Madsen, R.; Roberts, C.; Fraser-Reid, B. A Ready, Convergent Synthesis of the Heptasaccharide GPI Membrane Anchor of Rat Brain Thy-1 Glycoprotein. J. Am. Chem. Soc. 1993, 115, 7886–7887. [Google Scholar] [CrossRef]
- Baeschlin, D.K.; Chaperon, A.R.; Green, L.G.; Hahn, M.G.; Ince, S.J.; Ley, S.V. 1,2-Diacetals in Synthesis: Total Synthesis of a Glycosylphosphatidylinositol Anchor of Trypanosoma Brucei. Chem. Eur. J. 2000, 6, 172–186. [Google Scholar] [CrossRef]
- Liu, X.; Kwon, Y.-U.; Seeberger, P.H. Convergent Synthesis of a Fully Lipidated Glycosylphosphatidylinositol Anchor of Plasmodium Falciparum. J. Am. Chem. Soc. 2005, 127, 5004–5005. [Google Scholar] [CrossRef] [PubMed]
- Swarts, B.M.; Guo, Z. Synthesis of a Glycosylphosphatidylinositol Anchor Bearing Unsaturated Lipid Chains. J. Am. Chem. Soc. 2010, 132, 6648–6650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kihlberg, J.; Eichler, E.; Bundle, D.R. The Design and Synthesis of Antibody Binding Site Probes: Three Pentasaccharide Analogues of the Brucella a Antigen Prepared by Activation in Situ of Thioglycoside with Bromine. Carbohydr. Res. 1991, 211, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Nandan, S.R. Synthesis of Capuramycin. J. Org. Chem. 1994, 59, 281–283. [Google Scholar] [CrossRef]
- Leigh, D.A.; Smart, J.P.; Truscello, A.M. Intermolecular Aglycon Transfer of Ethyl 1-Thiorhamnopyranosides under Koenigs—Knorr and Helferich Glycosylation Conditions. Carbohydr. Res. 1995, 276, 417–424. [Google Scholar] [CrossRef]
- Belot, F.; Jacquinet, J.-C. Intermolecular Aglycon Transfer of a Phenyl 1-Thiogalactosaminide Derivative under Trichloroacetimidate Glycosylation Conditions. Carbohydr. Res. 1996, 290, 79–86. [Google Scholar] [CrossRef]
- Du, Y.; Lin, J.; Linhardt, R.J. Regioselective Synthesis of L-Idopyranuronic Acid Derivatives: Intermolecular Aglycon Transfer of Dithioacetal Under Standard Glycosylation Conditions. J. Carbohydr. Chem. 1997, 16, 1327–1344. [Google Scholar] [CrossRef]
- Yu, H.; Yu, B.; Wu, X.; Hui, Y.; Han, X. Synthesis of a Group of Diosgenyl Saponins with Combined Use of Glycosyl Trichloroacetimidate and Thioglycoside Donors. J. Chem. Soc. Perkin Trans. 1 2000, 1445–1453. [Google Scholar] [CrossRef]
- Zhu, T.; Boons, G.-J. Intermolecular Aglycon Transfer of Rthyl Thioglycoside Can be Prevented by Judicious Choice of Protecting Groups. Carbohydr. Res. 2000, 329, 709–715. [Google Scholar] [CrossRef]
- Sherman, A.A.; Yudina, O.N.; Mironov, Y.V.; Sukhova, E.V.; Shashkov, A.S.; Menshov, V.M.; Nifantiev, N.E. Study of Glycosylation with N-trichloroacetyl-D-Glucosamine Derivatives in the Syntheses of the Spacer-Armed Pentasaccharides Sialyl Lacto-N-Neotetraose and Sialyl Lacto-N-Tetraose, Their Fragments, and Aanalogues. Carbohydr. Res. 2001, 336, 13–46. [Google Scholar] [CrossRef]
- Cheshev, P.E.; Kononov, L.O.; Tsvetkov, Y.E.; Shashkov, A.S.; Nifantiev, N.E. Syntheses of α- and β-Glycosyl Donors with a Disaccharide β-D-Gal-(1→3)-D-GalNAc Backbone. Russ. J. Bioorg. Chem. 2002, 28, 419–429. [Google Scholar]
- Geurtsen, R.; Boons, G.-J. Chemoselective Glycosylations of Sterically Hindered Glycosyl Acceptors. Tetrahedron Lett. 2002, 43, 9429–9431. [Google Scholar] [CrossRef]
- Tanaka, H.; Adachi, M.; Takahashi, T. Efficient Synthesis of Core 2 Class Glycosyl Amino Acids by One-Pot Glycosylation Approach. Tetrahedron Lett. 2004, 45, 1433–1436. [Google Scholar] [CrossRef]
- Xue, J.; Khaja, S.D.; Locke, R.D.; Matta, K.L. A Concise Synthesis of N-Trichloroethoxycarbonyl Lactosamine Trichloroacetimidate Donor and its Application in the Synthesis of Gal(β1-4)GlcNAc(β1-3)L-Fuc(α-OAll). Synlett 2004, 2004, 861–865. [Google Scholar] [CrossRef]
- Codée, J.D.C.; Stubba, B.; Schiattarella, M.; Overkleeft, H.S.; van Boeckel, C.A.A.; van Boom, J.H.; van der Marel, G.A. A Modular Strategy toward the Synthesis of Heparin-like Oligosaccharides Using Monomeric Building Blocks in a Sequential Glycosylation Strategy. J. Am. Chem. Soc. 2005, 127, 3767–3773. [Google Scholar] [CrossRef]
- Sun, J.; Han, X.; Yu, B. Synthesis of Anemoclemoside B, the First Natural Product with an Open-Chain Cyclic Acetal Glycosidic Linkage. Org. Lett. 2005, 7, 1935–1938. [Google Scholar] [CrossRef]
- Li, Z.; Gildersleeve, J.C. An Armed-Disarmed Approach for Blocking Aglycon Transfer of Thioglycoside. Tetrahedron Lett. 2007, 48, 559–562. [Google Scholar] [CrossRef]
- Kato, M.; Hirai, G.; Sodeoka, M. Studies on the Selectivity between Glycosylation and Intermolecular Aglycone Transfer of Thioglucoside in Synthesis of Lactose Derivatives. Chem. Lett. 2011, 40, 877–879. [Google Scholar] [CrossRef]
- Yu, B.; Tao, H. Glycosyl Trifluoroacetimidates. Part 1: Preparation and Application as New Glycosyl Donors. Tetrahedron Lett. 2001, 42, 2405–2407. [Google Scholar] [CrossRef]
- Cai, S.; Yu, B. Efficient Sialylation with Phenyltrifluoroacetimidates as Leaving Groups. Org. Lett. 2003, 5, 3827–3830. [Google Scholar] [CrossRef]
- Tanaka, H.; Iwata, Y.; Takahashi, D.; Adachi, M.; Takahashi, T. Efficient Stereoselective Synthesis of γ-N-Glycosyl Asparagines by N-Glycosylation of Primary Amide Groups. J. Am. Chem. Soc. 2005, 127, 1630–1631. [Google Scholar] [PubMed]
- Yu, F.; Nguyen, H.M. Studies on the Selectivity Between Nickel-Catalyzed 1,2-cis-2-Amin Glycosylation of Hydroxyl Groups of Thioglycoside Acceptors with C(2)-Substituted Benzylidene N-Phenyl Trifluoroacetimidates and Intermolecular Aglycon Transfer of the Sulfide Group. J. Org. Chem. 2012, 77, 7330–7343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.-T.; Gildersleeve, J.C. Mechanistic Studies and Methods to Prevent Aglycon Transfer of Thioglycoside. J. Am. Chem. Soc. 2006, 128, 11612–11619. [Google Scholar] [PubMed]
- Sakuda, S.; Zhou, Z.-Y.; Yamada, Y. Structure of a Novel Disulfide of 2-(N-Acetylcysteinyl)amido-2-Deoxy-α-D-Glucopyran-Osyl-Myo-Inositol Produced by Streptomyces sp. Biosci. Biotechnol. Biochem. 1994, 58, 1347–1348. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Fahey, R.C. Mycothiol Biochemistry. Arch. Microbiol. 2002, 178, 388–394. [Google Scholar] [CrossRef]
- Newton, G.L.; Av-Gay, Y.; Fahey, R.C. A Novel Mycothiol-Dependent Detoxification Pathway in Mycobacteria Involving Mycothiol S-conjugate Amidase. Biochemistry 2000, 39, 10739. [Google Scholar]
- Ajayi, K.; Thakur, V.V.; Lapo, R.C.; Knapp, S. Intramolecular α-glucosaminidation: Synthesis of Mycothiol. Org. Lett. 2010, 12, 2630–2633. [Google Scholar]
- Chung, C.C.; Zulueta, M.M.; Padiyar, L.T.; Hung, S.C. Desymmetrization of 2,4,5,6-Tetra-O-Benzyl-D-Myo-Inositol for the Synthesis of Mycothiol. Org. Lett. 2011, 13, 5496–5499. [Google Scholar]
- Lee, S.; Rosazza, J.P. First Total Synthesis of Mycothiol and Mycothiol Disulfide. Org. Lett. 2004, 6, 365–368. [Google Scholar]
- Nicholas, G.M.; Kovác, P.; Bewley, C.A. Total Synthesis and Proof of Structure of Mycothiol Bimane. J. Am. Chem. Soc. 2002, 124, 3492–3493. [Google Scholar]
- Jardine, M.A.; Spies, H.S.; Nkambule, C.M.; Gammon, D.W.; Steenkamp, D.J. Synthesis of Mycothiol, 1D-1-O-(2-[N-acetyl-L-Cysteinyl]amino-2-Deoxy-α-D-Glucopyranosyl)-MyoInositol, Principal Low Molecular Mass Thiol in the Actinomycetes. Bioorg. Med. Chem. 2002, 10, 875–881. [Google Scholar] [CrossRef]
- McConnell, M.S.; Yu, F.; Nguyen, H.M. Nickel-Catalyzed α-glycosylation of C(1)-Hydroxyl D-Myo-Inositol: A Formal Synthesis of Mycothiol. Chem. Commun. 2013, 49, 4313–4315. [Google Scholar] [CrossRef]
- Hu, Y.-P.; Lin, S.-Y.; Huang, C.-Y.; Zulueta, M.M.L.; Liu, J.-Y.; Chang, W.; Hung, S.-C. Synthesis of 3-O-sulfonated Heparan Sulfate Octasaccharides that Inhibit the Herpes Simplex Virus Type 1 Host-Cell Interaction. Nat. Chem. 2011, 3, 557–563. [Google Scholar] [CrossRef]
- Goel, M.; Azev, V.N.; d’Alarcao, M. The Biological Activity of Structurally Defined Inositol Glycans. Future Med. Chem. 2009, 1, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Sanai, Y. Functional Roles of Glycosphingolipids in Signal Transduction via Lipid Rafts. Glycoconj. J. 2000, 17, 153–162. [Google Scholar] [CrossRef]
- Paulick, M.G.; Forstner, M.B.; Groves, J.T.; Bertozzi, C.R. A Chemical Approach to Unraveling the Biological Function of the Glycosylphosphatidylinositol Anchor. Proc. Natl. Acad. Sci. USA 2007, 104, 20332–20337. [Google Scholar] [CrossRef] [Green Version]
- McConnell, M.S.; Mensah, E.A.; Nguyen, H.M. Stereoselective α-glycosylation of C(6)-Hydroxyl Myo-Inositols via Nickel Catalysis-Application to the Synthesis of GPI Anchor Pseudo-Oligosaccharides. Carbohydr. Res. 2013, 381, 146–152. [Google Scholar] [CrossRef]
- Tsai, Y.-H.; Götze, S.; Vilotijevic, I.; Grube, M.; Silva, D.V.; Seeberger, P.H. A General and Convergent Synthesis of Diverse Glycosylphosphatidylinositol Glycolipids. Chem. Sci. 2013, 4, 468–481. [Google Scholar] [CrossRef]
- Swarts, B.M.; Guo, Z. Chapter 4—Chemical Synthesis of Glycosylphosphatidylinositol Anchors. In Advances in Carbohydrate Chemistry and Biochemistry; Horton, D., Ed.; Academic Press: Cambridge, MA, USA, 2012; pp. 137–219. [Google Scholar]
- Homans, S.W.; Ferguson, M.A.J.; Dwek, R.A.; Rademacher, T.W.; Anand, R.; Williams, A.F. Complete Structure of the Glycosyl Phosphatidylinositol Membrane Anchor of Rat Brain Thy-1 Glycoprotein. Nature 1988, 333, 269–272. [Google Scholar] [CrossRef]
- Pratt, M.R.; Bertozzi, C.R. Synthetic Glycopeptides and Glycoproteins as Tools for Biology. Chem. Soc. Rev. 2005, 34, 58–68. [Google Scholar] [CrossRef]
- Slovin, S.F.; Ragupathi, G.; Musselli, C.; Olkiewicz, K.; Verbel, D.; Kuduk, S.D.; Schwarz, J.B.; Sames, D.; Danishefsky, S.; Livingston, P.O.; et al. Fully Synthetic Carbohydrate-Based Vaccines in Biochemically Relapsed Prostate Cancer: Clinical Trial Results with α-N-Acetylgalactosamine-O-Serine/Threonine Conjugate Vaccine. J. Clin. Oncol. 2003, 21, 4292–4298. [Google Scholar] [CrossRef]
- Springer, G.F. Immunoreactive T and Tn Epitopes in Cancer Diagnosis, Prognosis, and Immunotherapy. J. Mol. Med. 1997, 75, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Dziadek, S.; Hobel, A.; Schmitt, E.; Kunz, H. A Fully Synthetic Vaccine Consisting of a Tumor-Associated Glycopeptide Antigen and a T-Cell Epitope for the Induction of a Highly Specific Humoral Immune Response. Angew. Chem. Int. Ed. 2005, 44, 7630–7635. [Google Scholar] [CrossRef] [PubMed]
- Kuduk, S.D.; Schwarz, J.B.; Chen, X.-T.; Glunz, P.W.; Sames, D.; Ragupathi, G.; Livingston, P.O.; Danishefsky, S.J. Synthetic and Immunological Studies on Clustered Modes of Mucin-Related TN and TF O-Linked Antigens: The Preparation of a Glycopeptide-Based Vaccine for Clinical Trials against Prostate Cancer. J. Am. Chem. Soc. 1998, 120, 12474–12485. [Google Scholar] [CrossRef]
- Yu, F.; McConnell, M.S.; Nguyen, H.M. Scalable Synthesis of Fmoc-Protected GalNAc-Threonine Amino Acid and TN Antigen via Nickel Catalysis. Org. Lett. 2015, 17, 2018–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sletten, E.T.; Ramadugu, S.K.; Nguyen, H.M. Utilization of Bench-Stable and Readily Available Nickel(II) Triflate for Access to 1,2-cis-2-Aminoglycoside. Carbohydr. Res. 2016, 435, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Samala, G.; Sletten, E.T.; Stockdill, J.L.; Nguyen, H.M. Facile Triflic Acid-Catalyzed α-1,2-cis-Thio Glycosylations: Scope and Application to the Synthesis of S-linked Oligosaccharides, Glycolipids, Sublancin Glycopeptides, and TN/TF Antigens. Chem. Sci. 2019, 10, 10475–10480. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Sun, G.; Yao, W.; Zhu, Y.; Wang, R.; Cai, L.; Liu, K.; Zhang, Q.; Liu, X.-W.; Wan, Q. 3-Aminodeoxypyranoses in Glycosylation: Diversity-Oriented Synthesis and Assembly in Oligosaccharides. Angew. Chem. Int. Ed. 2017, 56, 5227–5231. [Google Scholar] [CrossRef]
- Zeng, J.; Sun, G.; Wang, R.; Zhang, S.; Teng, S.; Liao, Z.; Meng, L.; Wan, Q. Gold-Catalyzed Diversified Synthesis of 3-Aminosugar Analogues of Digitoxin and Digoxin. Org. Chem. Front. 2017, 4, 2450–2454. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, R.; Yao, W.; Zhang, S.; Sun, G.; Liao, Z.; Meng, L.; Wan, Q. Diversified Synthesis and α-Selective Glycosylation of 3-Amino-2,3,6-Trideoxy Sugars. Org. Chem. Front. 2018, 5, 3391–3395. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, R.; Zhang, S.; Fang, J.; Liu, S.; Sun, G.; Xu, B.; Xiao, Y.; Fu, D.; Zhang, W.; et al. Hydrogen-Bonding-Assisted Exogenous Nucleophilic Reagent Effect for β-Selective Glycosylation of Rare 3-Amino Sugars. J. Am. Chem. Soc. 2019, 141, 8509–8515. [Google Scholar] [CrossRef]
- Aoki, M.; Shirai, H.; Nakayama, N.; Itezono, Y.; Mori, M.; Satoh, T.; Ohshima, S.; Watanabe, J.; Yokose, K.; Seto, H. Structural Studies on Avidinorubicin, a Novel Anthracycline with Platelet Aggregation Inhibitory Activity. J. Antibiot. 1991, 44, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Hoang, K.M.; Lees, N.R.; Herzon, S.B. General Method for the Synthesis of α- or β-Deoxyaminoglycoside Bearing Basic Nitrogen. J. Am. Chem. Soc. 2021, 143, 2777–2783. [Google Scholar] [CrossRef] [PubMed]
- Hoang, K.M.; Lees, N.R.; Herzon, S.B. Programmable Synthesis of 2-Deoxyglycoside. J. Am. Chem. Soc. 2019, 141, 8098–8103. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.R.; Rychnovsky, S.D. Generation, Stability, and Utility of Lithium 4,4′-Di-Tert-Butylbiphenylide (LiDB). J. Org. Chem. 2016, 81, 10707–10714. [Google Scholar] [CrossRef]
- Cohen, T.; Lin, M.T. Two-Flask Preparation of Alpha.-Lithio Cyclic Ethers from Gamma.- and .Delta.-Lactones. Reductive Lithiation as a Route, via Radical Intermediates, to Axial 2-Lithiotetrahydropyrans and Their Equilibration to the Equatorial Isomers. J. Am. Chem. Soc. 1984, 106, 1130–1131. [Google Scholar] [CrossRef]
- Cohen, T.; Bhupathy, M. Organoalkali Compounds by Radical Anion Induced Reductive Metalation of Phenyl Thioethers. Acc. Chem. Res. 1989, 22, 152–161. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Mickus, D.E. Preparation of 2-Lithiotetrahydropyrans: Kinetic and Thermodynamic Generation of Alkyllithium Reagents. Tetrahedron Lett. 1989, 30, 3011–3014. [Google Scholar] [CrossRef]
- Baryal, K.N.; Zhu, D.; Li, X.; Zhu, J. Umpolung Reactivity in the Stereoselective Synthesis of S-linked 2-Deoxyglycoside. Angew. Chem. Int. Ed. 2013, 52, 8012–8016. [Google Scholar] [CrossRef]
- Kyasa, S.; Meier, R.N.; Pardini, R.A.; Truttmann, T.K.; Kuwata, K.T.; Dussault, P.H. Synthesis of Ethers via Reaction of Carbanions and Monoperoxyacetals. J. Org. Chem. 2015, 80, 12100–12114. [Google Scholar] [CrossRef]
- Stich, H.F.; Stich, W.; Acton, A.B. Mutagenicity of Fecal Extracts from Carnivorous and Herbivorous Animals. Mutat. Res. 1980, 78, 105–112. [Google Scholar] [CrossRef]
- Lehn, J.M.; Wipff, G. Stereoelectronic Properties, Stereospecificity, and Stabilization of α-Oxa and α-Thia Carbanions. J. Am. Chem. Soc. 1976, 98, 7498–7505. [Google Scholar] [CrossRef]
- Liu, D.M.; Wang, H.L.; Lei, J.C.; Zhou, X.Y.; Yang, J.S. A Highly α-Stereoselective Sialylation Method Using 4-O-4-Nitropicoloyl Thiosialoside Donor. Eur. J. Org. Chem. 2020, 2020, 575–585. [Google Scholar] [CrossRef]
- Yu, C.S.; Niikura, K.; Lin, C.C.; Wong, C.H. The Thioglycoside and Glycosyl Phosphite of 5-Azido Sialic Acid: Excellent Donors for the α-Glycosylation of Primary Hydroxy Groups. Angew. Chem. Int. Ed. 2001, 40, 2900–2903. [Google Scholar] [CrossRef]
- Mandhapati, A.R.; Rajender, S.; Shaw, J.; Crich, D. The Isothiocyanato Moiety: An Ideal Protecting Group for the Stereoselective Synthesis of Sialic Acid glycoside and Subsequent Diversification. Angew. Chem. Int. Ed. 2015, 54, 1275–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, F.; Ishiwata, A.; Ito, Y. Stereodivergent Mannosylation Using 2- O-(ortho-Tosylamido)benzyl Group. Org. Lett. 2018, 20, 4833–4837. [Google Scholar] [CrossRef]
- Escopy, S.; Geringer, S.A.; de Meo, C. Combined Effect of the Picoloyl Protecting Group and Triflic Acid in Sialylation. Org. Lett. 2017, 19, 2638–2641. [Google Scholar] [CrossRef]
- Jones, B.; Behm, A.; Shadrick, M.; Geringer, S.A.; Escopy, S.; Lohman, M.; de Meo, C. Comparative Study on the Effects of Picoloyl Groups in Sialylations Based on Their Substitution Pattern. J. Org. Chem. 2019, 84, 15052–15062. [Google Scholar] [CrossRef]
- Asressu, K.H.; Chang, C.W.; Lam, S.; Wang, C.C. Donor-Reactivity-Controlled Sialylation Reactions. Eur. J. Org. Chem. 2021, 2021, 4525–4530. [Google Scholar] [CrossRef]
- Hayashi, T.; Axer, A.; Kehr, G.; Bergander, K.; Gilmour, R. Halogen-Directed Chemical Sialylation: Pseudo-Stereodivergent Access to Marine Ganglioside Epitopes. Chem. Sci. 2020, 11, 6527–6531. [Google Scholar] [CrossRef] [Green Version]
- Kurimoto, K.; Yamamura, H.; Miyagawa, A. Sialylation with Systematically Protected Allyl Galactoside Acceptors Using Sialyl Phosphate Donors. Tetrahedron Lett. 2016, 57, 1442–1445. [Google Scholar] [CrossRef]
- Wang, J.; Lou, Q.; Rong, J.; Yang, Y. Gold(I)-Promoted α-selective Sialylation of Glycosyl ortho-Hexynylbenzoates for the Latent-Active Ssynthesis of Oligosialic Acids. Org. Biomol. Chem. 2019, 17, 6580–6584. [Google Scholar] [CrossRef]
- Boons, G.J.; Demchenko, A.V. Recent Advances in O-sialylation. Chem. Rev. 2000, 100, 4539–4565. [Google Scholar] [CrossRef]
- Sun, B. Recent Developments of Highly Selective Sialylation. Curr. Org. Chem. 2016, 20, 1465–1476. [Google Scholar] [CrossRef]
- Vibhute, A.M.; Komura, N.; Tanaka, H.N.; Imamura, A.; Ando, H. Advanced Chemical Methods for Stereoselective Sialylation and Their Applications in Sialoglycan Syntheses. Chem. Rec. 2021, 21, 3194–3223. [Google Scholar] [CrossRef]
Figure 1.
Selected Natural products and pharmaceuticals containing amino sugars. (A) Selected medicine containing aminoglycosides sub-unit, (B) Selected natural products containing aminoglycosides sub-unit, (C) Some other important amino sugars.
Figure 1.
Selected Natural products and pharmaceuticals containing amino sugars. (A) Selected medicine containing aminoglycosides sub-unit, (B) Selected natural products containing aminoglycosides sub-unit, (C) Some other important amino sugars.

Figure 2.
Structure of 1,2-cis- and 1,2-trans-aminosugars.

Scheme 1.
The strategy for the formation of glycoside of 2-amino sugars (A) Glycosylation of 2-amino sugars with a neighboring participating group at C(2) position, (B) Glycosylation of 2-amino sugars without a neighboring participating group at C(2) position.
Scheme 1.
The strategy for the formation of glycoside of 2-amino sugars (A) Glycosylation of 2-amino sugars with a neighboring participating group at C(2) position, (B) Glycosylation of 2-amino sugars without a neighboring participating group at C(2) position.

Scheme 2.
Glycosylation of DNs-protected donor 11 and 12 with various nucleophiles.

Figure 3.
Proposed mechanism for glycosylation of 11.

Scheme 3.
DNs-directed SN2-like glycosylation of selenoglycosaminosides.

Scheme 4.
Synthesis of α-disaccharides by in situ activation of anomeric OH in 34 and 35.

Scheme 5.
Glycosylation with benzoylated and benzylated galactosyl nitrate.

Scheme 6.
Glycosylation with 2-azidoglucosyl nitrate donor 59.

Scheme 7.
The synthesis of glycosylphosphatidylinositol (GPI) core by Guo’s group.

Scheme 8.
The synthesis of the TN-antigen building block.

Scheme 9.
The construction of a 2-amino-α-glycosidic bond by using conformation-locked acceptors.

Scheme 10.
The glycosylation of glycosyl donors 75 and 76 with glycosyl acceptors 77 and 52 in the presence of thiophene.
Scheme 10.
The glycosylation of glycosyl donors 75 and 76 with glycosyl acceptors 77 and 52 in the presence of thiophene.

Scheme 11.
The formation of TN-antigen building blocks.

Scheme 12.
The synthesis and protecting group manipulation of 3-fluoro TN antigen analogues.

Scheme 13.
The results of glycosylation.

Scheme 14.
The assembly of an α-glucosazide tetrasaccharide using MPF-mediated glycosylation.

Scheme 15.
The assembly of a Pel fragment 116.

Scheme 16.
Michael-type addition of alcohols to 2-nitro-D-galactal derivatives.

Scheme 17.
Stereoselective glycosylation of 117 using P4-t-Bu as superbase catalyst.

Scheme 18.
Cinchona-thiourea-catalyzed glycosylation of 2-nitrogalactals 117.

Scheme 19.
α-Selective glycosylation using 2-nitrogalactal 117.

Scheme 20.
α-Selective glycosylation with 2-nitroglucal 162.

Scheme 21.
Thiourea 170-catalyzed glycosylation of 2-nitroglycal 166.

Scheme 22.
Thiourea 170-catalyzed glycosylation of 2-nitrogalactal 175.

Scheme 23.
NHC-catalyzed stereoselective glycosylation of 2-nitrogalactals with alcohols and phenol.
Scheme 23.
NHC-catalyzed stereoselective glycosylation of 2-nitrogalactals with alcohols and phenol.

Scheme 24.
Ring-fused oxazolidinone for the stereoselective formation of α-linked glycoside.

Scheme 25.
The formation of α-linked serine and threonine derivatives.

Scheme 26.
The glycosidation of donor 204 under BSP/Tf2O activation conditions.

Scheme 27.
Glycosylation reactions under two different conditions.

Scheme 28.
Stereoselectivities of glycosylation reactions using the novel glycosyl donor.

Scheme 29.
The preparation of trisaccharide 236.

Scheme 30.
The glycosylation of 237 with acceptors by pre-activation in the presence of TTBP.

Scheme 31.
The glycosylation of 237 with acceptors by pre-activation in the absence of TTBP.

Scheme 32.
Nickel-catalyzed stereoselective formation of 1,2-cis-aminoglycoside.

Scheme 33.
Comparison between Lewis acids and nickel catalyst.

Scheme 34.
Substrate scope of nickel-catalyzed 1,2-cis-amino glycosylation.

Scheme 35.
α-Selective coupling with D-galactosamine trichloroacetimidate.

Scheme 36.
α-Selective glycosylation of disaccharide acceptors.

Scheme 37.
α-Selective glycosylation of glucuronic acid acceptors.

Scheme 38.
α-Selective glycosylation of inositol acceptor.

Figure 4.
The proposed mechanism for nickel-catalyzed 1,2-cis-2-amino glycosylation.

Scheme 39.
Coupling of various thioglycosides with N-phenyl trifluoroacetimidate donors.

Scheme 40.
Glycosylation with C(2)-azido imidate donor.

Scheme 41.
(a) The synthesis of trisaccharide 349; (b) The synthesis of trisaccharide 352.

Figure 5.
The structure of mycothiol.

Scheme 42.
The synthesis of core pseudosaccharides of mycothiol.

Scheme 43.
The formation of mycothiol.

Scheme 44.
The formation of the GPI anchor core.

Figure 6.
The structure of TN, TF and STN antigens.

Scheme 45.
Synthesis of TN antigen core via nickel-catalyzed glycosylation.

Scheme 46.
Gram-scale synthesis of TN antigen.

Scheme 47.
H-bond-stabilized oxyphosphonium species for β-glycosylation.

Scheme 48.
Substrate scope.

Scheme 49.
Synthesis of the trisaccharide unit of avidinorubicin.

Scheme 50.
Anomeric anion approach to aminoglycoside.

Scheme 51.
The synthesis of α-aminoglycoside.

Scheme 52.
The synthesis of β-aminoglycoside.

Scheme 53.
The synthesis of 4-aminoglycoside.

Scheme 54.
Design of potential sialyl donor for α-glycosylation.

Figure 7.
Glycosylation between donors 431–433 and acceptor 434.

Scheme 55.
The synthesis of protected trisaccharide 447.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, J.; Xie, D.; Ma, X. Recent Advances in Chemical Synthesis of Amino Sugars. Molecules 2023, 28, 4724. https://doi.org/10.3390/molecules28124724
AMA Style
Yang J, Xie D, Ma X. Recent Advances in Chemical Synthesis of Amino Sugars. Molecules. 2023; 28(12):4724. https://doi.org/10.3390/molecules28124724
Chicago/Turabian StyleYang, Jian, Demeng Xie, and Xiaofeng Ma. 2023. "Recent Advances in Chemical Synthesis of Amino Sugars" Molecules 28, no. 12: 4724. https://doi.org/10.3390/molecules28124724