 Bert Malengier-Devlies
Bert Malengier-Devlies Mieke Metzemaekers
Mieke Metzemaekers Carine Wouters
Carine Wouters Paul Proost
Paul Proost Patrick Matthys
Patrick Matthys- 1Department of Microbiology, Immunology and Transplantation, Laboratory of Immunobiology, Rega Institute for Medical Research, KU Leuven, Leuven, Belgium
- 2Department of Microbiology, Immunology and Transplantation, Laboratory of Molecular Immunology, Rega Institute for Medical Research, KU Leuven, Leuven, Belgium
- 3Division of Pediatric Rheumatology, University Hospitals Leuven, Leuven, Belgium
- 4European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases (RITA) at University Hospital Leuven, Leuven, Belgium
Neutrophils are key pathogen exterminators of the innate immune system endowed with oxidative and non-oxidative defense mechanisms. More recently, a more complex role for neutrophils as decision shaping cells that instruct other leukocytes to fine-tune innate and adaptive immune responses has come into view. Under homeostatic conditions, neutrophils are short-lived cells that are continuously released from the bone marrow. Their development starts with undifferentiated hematopoietic stem cells that pass through different immature subtypes to eventually become fully equipped, mature neutrophils capable of launching fast and robust immune responses. During severe (systemic) inflammation, there is an increased need for neutrophils. The hematopoietic system rapidly adapts to this increased demand by switching from steady-state blood cell production to emergency granulopoiesis. During emergency granulopoiesis, the de novo production of neutrophils by the bone marrow and at extramedullary sites is augmented, while additional mature neutrophils are rapidly released from the marginated pools. Although neutrophils are indispensable for host protection against microorganisms, excessive activation causes tissue damage in neutrophil-rich diseases. Therefore, tight regulation of neutrophil homeostasis is imperative. In this review, we discuss the kinetics of neutrophil ontogenesis in homeostatic conditions and during emergency myelopoiesis and provide an overview of the different molecular players involved in this regulation. We substantiate this review with the example of an autoinflammatory disease, i.e. systemic juvenile idiopathic arthritis.
Introduction
Neutrophils are the most abundant leukocytes in human blood and protect our bodies from potentially harmful agents (1, 2). The importance of these cells is demonstrated in patients with neutropenia, leukocyte adhesion deficiency, or chronic granulomatous disease who are prone to developing (fatal) microbial infections (3). Traditionally, neutrophils were considered to be homogeneous, simple, and short-lived innate phagocytes mounting rapid - but largely non-specific - antibacterial and antifungal responses. However, discoveries in recent years have revealed the complexity of neutrophil functions including phagocytosis, degranulation, ROS production and NET formation (1, 4, 5). In addition, neutrophils interact with other leukocytes through the production of alarmins (such as S100A8/A9 and S100A12), cytokines and chemokines. Neutrophils have both disease-promoting and disease-limiting properties, indicative of the existence of different neutrophil subsets with unique immunomodulatory functions (6–9). Indeed, the existence of neutrophil subsets with transcriptional, functional, and phenotypic heterogeneity has recently emerged in both humans and mice (10). The production and storage of new neutrophils are tightly regulated. Figure 1 summarises the different neutrophil regulatory mechanisms that include: the formation of new neutrophils or ‘granulopoiesis’ in the bone marrow (BM), the release of new neutrophils into the circulation, the storage of neutrophils outside the BM (marginated pool), the infiltration of neutrophils into sites of inflammation (tissue pool) and eventually the clearance of (aged) neutrophils (7, 11). Under steady-state conditions, neutrophils are produced in the BM at a rate of 1-2 x 1011 cells/day in humans and 1 x 107 cells/day in mice (12, 13). However, during an excessive inflammatory immune response, when there is a high demand for new neutrophils, the life span of neutrophils may increase up to 7 days and the production of new neutrophils may increase tenfold (11, 14). During such excessive neutrophil production - often referred to as ‘emergency granulopoiesis’ - the generation of new cells may additionally take place outside the bone marrow (BM) in a process called ‘extramedullary myelopoiesis’ (15, 16). Diverse autoimmune and autoinflammatory diseases are hallmarked by emergency granulopoiesis. In this review, we provide an overview of the mechanisms and molecules that regulate homeostatic and emergency granulopoiesis. We place special emphasis on neutrophils and cytokines in systemic juvenile idiopathic arthritis (sJIA), which is a rare but severe multifactorial autoinflammatory disease characterised by expansion of neutrophils.
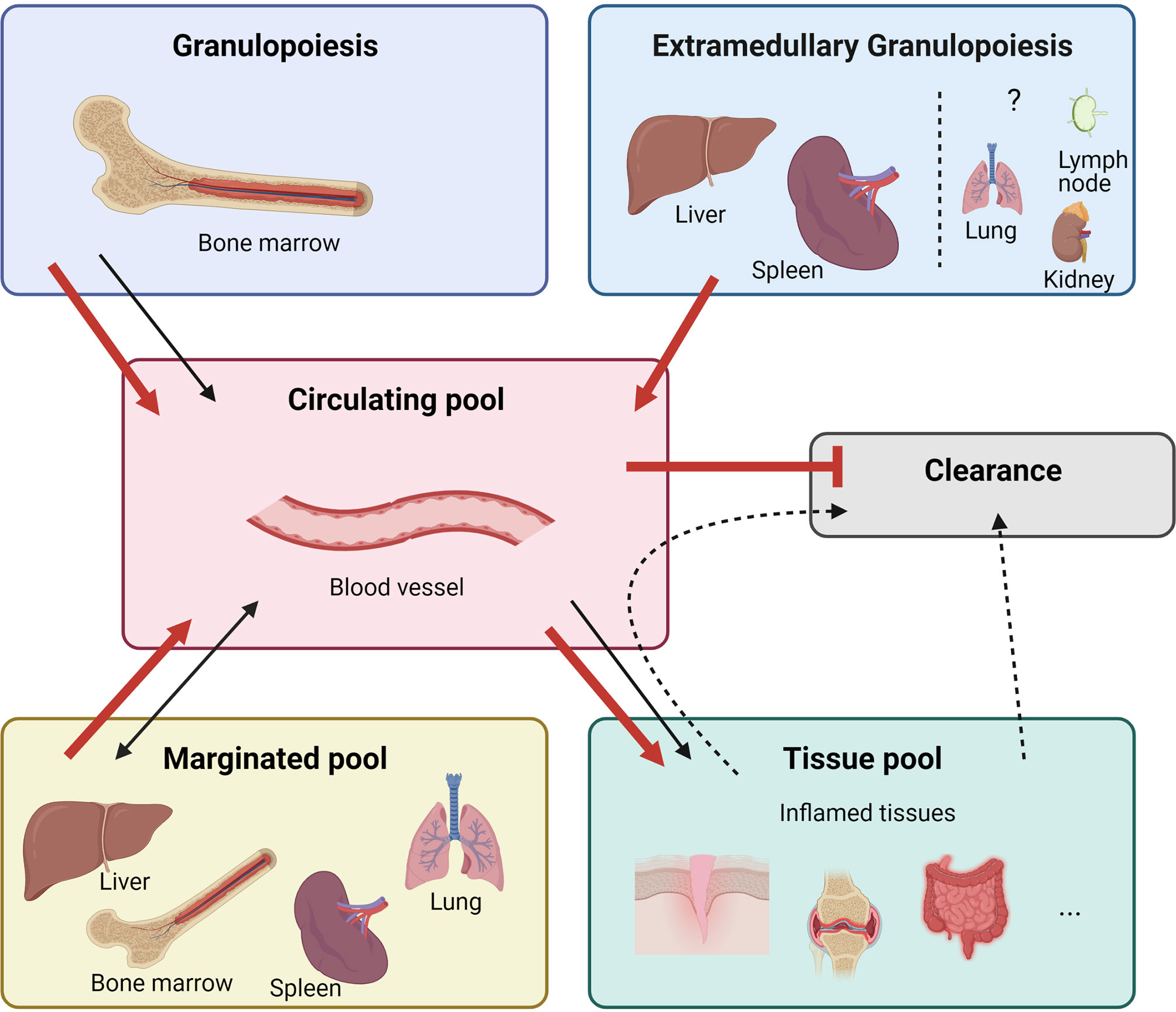
Figure 1 Neutrophil homeostasis is tightly regulated. In homeostatic conditions (black arrows) neutrophils mature in the bone marrow from undifferentiated hematopoietic stem cells (HSCs), replenishing the mitotic stem cell pool that gives rise to mature neutrophils via different immature stages of myeloblast, promyelocyte, myelocyte, metamyelocyte, immature band cells, and mature neutrophils. These neutrophils are stored in the bone marrow upon release before entering the blood circulation. Different organs store a pool of neutrophils, referred to as marginated pool, and are forming a reservoir of mature neutrophils. In response to inflammation, neutrophils can exit the bloodstream and enter the tissue pool. Senescent neutrophils are cleared in the bone marrow by stromal macrophages or by tissue-resident macrophages in the periphery. During excessive inflammation (red arrows), neutrophils are massively attracted to the site of inflammation and their clearance is decreased by increasing their lifespan. Demanding the high need for neutrophils, increased numbers of neutrophils are released from the bone marrow. Furthermore, neutrophils are formed and released from extramedullary sites. Cytokine stimulation, including epinephrine, can additionally quickly release neutrophils from the marginated pool.
Regulation of Neutrophil Homeostasis and Major Functions of Neutrophils
Granulopoiesis: A Process That Takes More Than 10 Days
The BM is the main source of new neutrophils. Neutrophils that mature in the BM can be subdivided into three pools with increasingly restricted proliferation potentials, i.e. the stem cell pool, the mitotic pool, and the post-mitotic pool. The hematopoietic stem cells (HSC) of the stem cell pool give rise to the granulocyte-macrophage progenitor (GMP) cells that gradually mature under influence of different cytokines or growth factors into mature neutrophils. Complete neutrophil maturation takes more than 10 days (17, 18).
HSCs localize in dedicated BM niches filled by perivascular cells that express a membrane-bound form of stem cell factor as well as the chemokine CXCL12 (also known as stromal cell-derived factor 1 or SDF-1), which are the ligands for the stem cell antigen CD117 (C-kit) and CXC chemokine receptor 4 (CXCR-4), respectively (19, 20). Two models are proposed that describe the differentiation of these HSCs. In the first classical or hierarchical model, HSCs give rise to committed common myeloid progenitors (CMP) that eventually give rise to GMPs. During the differentiation, their capacity to give rise to other cell types is progressively lost (21–23). In the alternative model, cells rather have a mixed-lineage potential with transcriptional and functional heterogeneity (21). The absence of oligopotent intermediates with mixed cell markers, having a limited differentiation potential, is favouring this second model (24–26). Here, the CMP compartment contains predestined subpopulations that are transcriptionally primed towards becoming either erythrocytes, megakaryocytes, dendritic cells (DCs), monocytes, neutrophils, eosinophils, or basophils (26–28).
The mitotic pool includes promyelocytes and myelocytes. Finally, the most differentiated non-dividing cells include both the immature neutrophils and the mature neutrophils. The different neutrophil precursors can be distinguished microscopically. Promyelocytes are defined as large cells with an oval nucleus and dark cytoplasm. Myelocytes have a less dense cytoplasm and a round-shaped nucleus. Metamyelocytes, band cells, and mature neutrophils are relatively small cells and have a clear cytoplasm. In humans, metamyelocytes and band cells have a kidney-like nucleus whereas in mice these cells have a doughnut-like, band-shaped nucleus. Finally, mature neutrophils are characterised by their segmented nuclei (29–34). The different neutrophil precursors are shown in Figure 2.
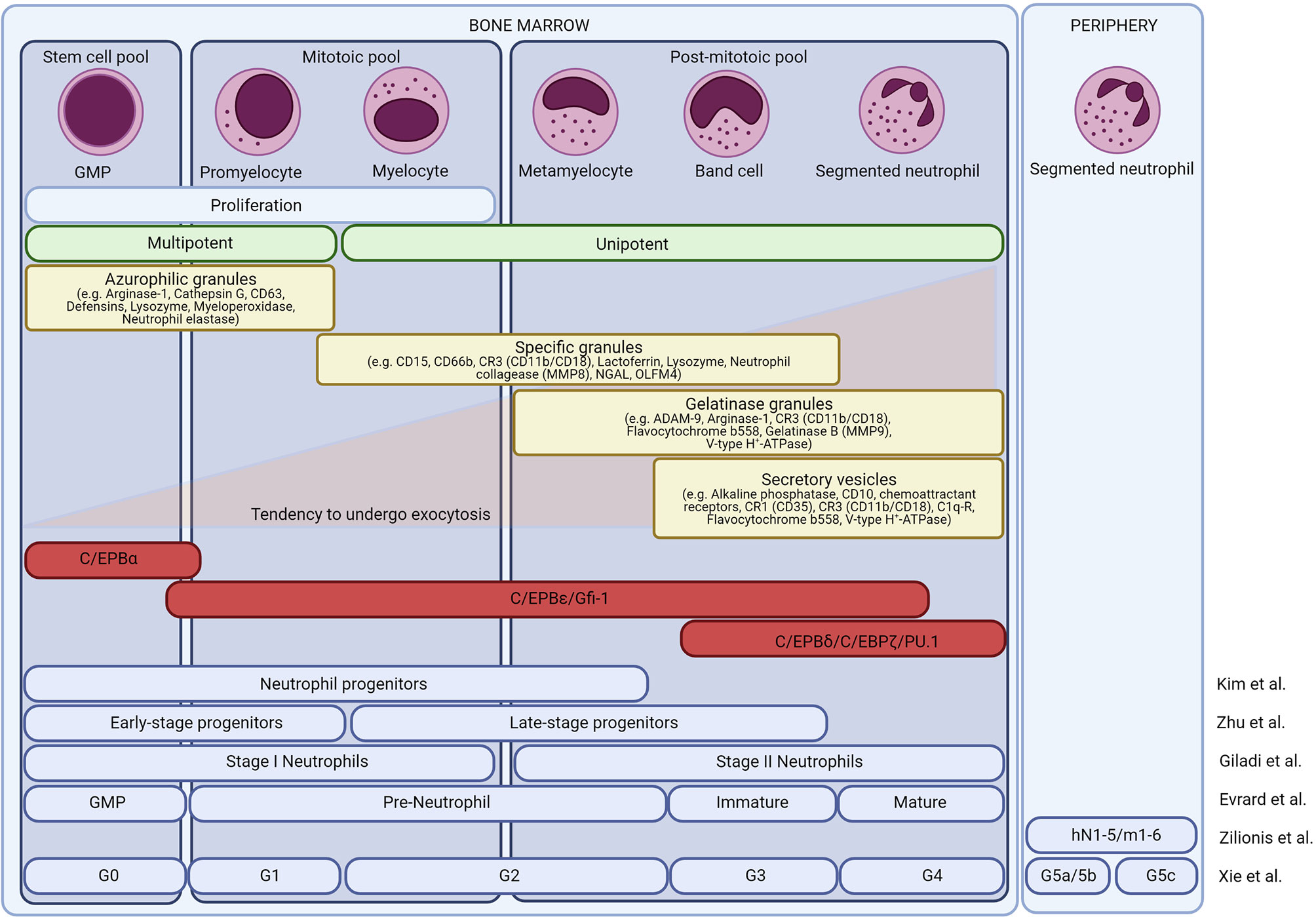
Figure 2 Neutrophil development in the bone marrow. Overview of the different neutrophil subsets in the bone marrow. Morphologically, neutrophils are divided into granulocyte-monocyte progenitor cells (GMPs), promyelocytes, myelocytes, metamyelocytes, band neutrophils, and mature neutrophils. Based on their proliferative and differentiation capacity, the cells are divided into a stem cell pool, a mitotic pool, and a post-mitotic pool. Neutrophil maturation is associated with changes in transcription factors and granule protein expression. At least four types of granules are formed in the neutrophils, each obtaining a unique set of effector molecules. The different granules are released hierarchically, opposite to their formation. Recently, single-cell sequencing or CyTOF reassessed the different neutrophil subsets and different groups have proposed a new neutrophil nomenclature.
The differentiation of neutrophils requires dynamic changes in the activity of specific transcription factors. The main transcription factors driving granulopoiesis are CCAAT/enhancer-binding protein (C/EBP) α and PU.1 (Figure 3). High expression of C/EBPα or PU.1 is associated with commitment to the granulocyte or monocyte lineage, respectively. Runx1 and lymphoid enhancer-binding factor 1 (Lef1) are responsible for regulating the expression of C/EBPα with defects in their activities resulting in a neutrophil maturational block (33, 35, 36). C/EBPα itself negatively regulates the expression of cMyc. Consistently, C/EBPα mutant mice have an early block in granulocyte differentiation (37–39). Another major transcription factor involved in neutrophil development is Krüppel-like factor 5 (KLF5), which is mainly active during early developmental stages and controls neutrophil production at the expanse of eosinophils (40). In addition, growth factor independent-1 (Gfi-1) also drives the initial neutrophil development and represses the monocyte-promoting transcription factors PU.1 and Irf8 (26, 41–47). Mutations in Gfi-1 block neutrophil maturation at the promyelocyte stage (33, 36, 46). The expression of Gfi-1 and C/EBPα decreases from the myeloblast stage and is associated with an increased expression of C/EBPϵ that peaks at the myelocyte stage. C/EBPϵ regulates the transition from promyelocytes to myelocytes and represses genes involved in cell cycling. The deletion of C/EBPϵ leads to neutrophil progenitor arrest and neutropenia (26, 48–51). Further myelocyte maturation is driven by an upregulation of C/EBPβ, C/EBPγ, C/EBPδ, and C/EBPξ.
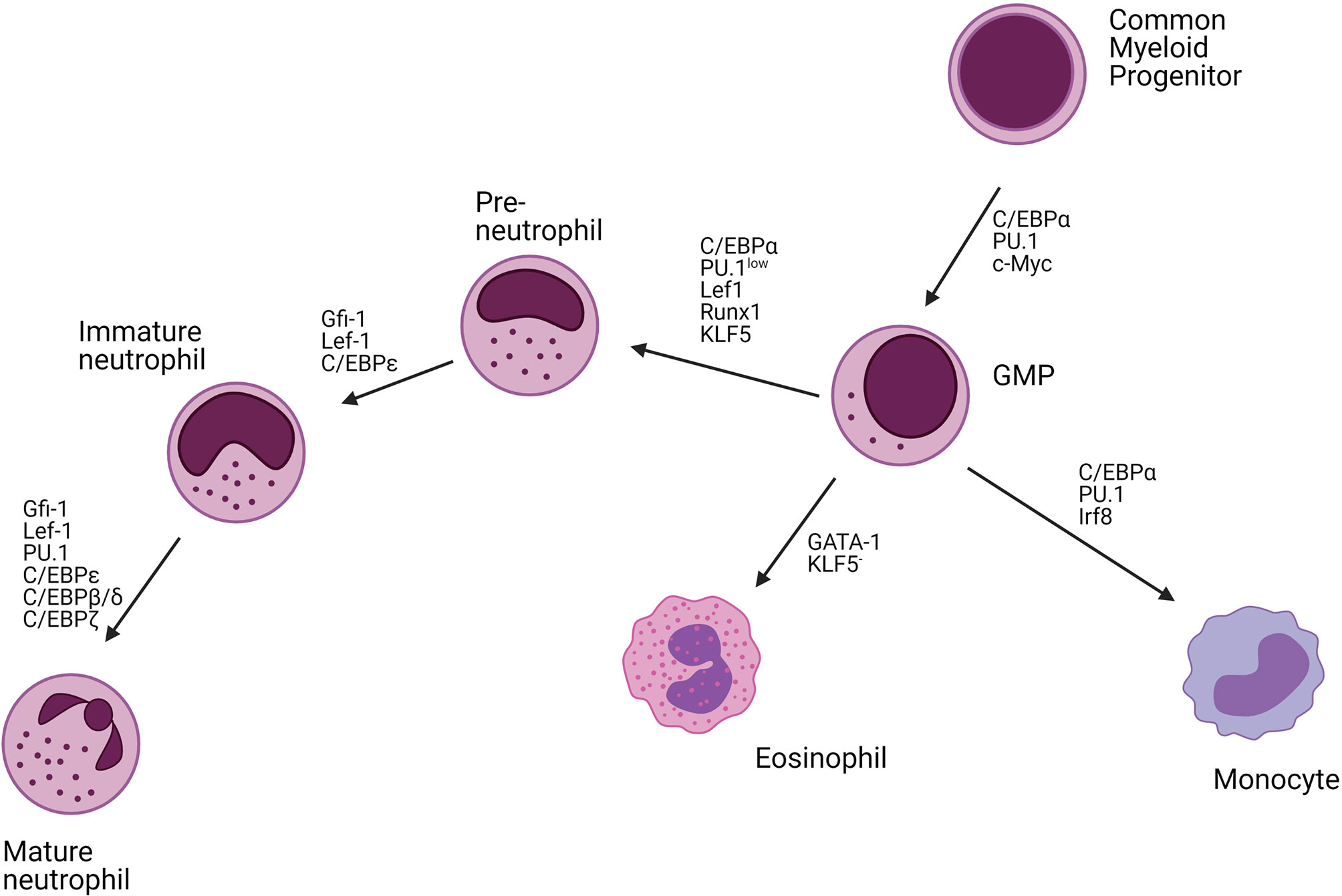
Figure 3 Transcription factors regulating neutrophil differentiation. Neutrophils mature from hematopoietic stem cells (HSCs) in the bone marrow (BM). These HSCs are self-reviewal and can differentiate into all immune cells. The common myeloid progenitor (CMP) cells give rise to the myeloid lineage including neutrophils, monocytes, and eosinophils. C/EBPα is the main transcription factor driving myeloid differentiation. Together with PU.1, and c-Myc, C/EBPα drives granulocyte-monocyte progenitor cell (GMP) differentiation. High levels of C/EBPα, PU.1, and Irf8 further drive monocyte development, high levels of GATA-1 eosinophil development, whereas C/EBPα, Lef1, Runx1, KLF5 together with low expression of PU.1 enhance neutrophils development. Neutrophil maturation is further driven by Gfi-1, Lef-1, C/EBP transcription factors (C/EBPϵ, C/EBPβ, C/EBPδ, C/EBPζ), and PU.1. C/EBP, CCAAT/enhancer-binding protein; Irf8, Interferon regulatory factor 8; Lef1, Lymphoid enhancer-binding factor 1; Runx1, Runt-related transcription factor 1; KLF5, Krüppel Like Factor 5; GFI1, Growth Factor Independent 1 Transcriptional Repressor.
The different maturational stages are accompanied by the sequential formation of primary (azurophilic), secondary (specific), tertiary (gelatinase), and secretory granules (34). Primary granules are mainly formed during the myeloblast stage (GMP, promyelocyte) and the main granule proteins include myeloperoxidase (MPO), α-defensins, bactericidal/permeability-increasing protein (BPI), and distinct serine proteinases such as elastase or cathepsin G (Figure 2). The main secondary granule proteins are lactoferrin (LTF) and lysozyme and are mainly formed during the myelocyte stages (myelocyte, metamyelocyte). Tertiary granules are rich in gelatinases and are formed during the band cell stage (banded immature neutrophils). Secretory vesicles are mainly generated during the final stages of neutrophil maturation and contain early activation-related proteins that facilitate neutrophil adhesion and migration. Upon neutrophil activation, the different granules are hierarchically released according to the “formed-first-released-last model” (52).
Novel insights on granulopoiesis were recently provided by innovative approaches such as single-cell RNA sequencing (scRNAseq) and mass cytometry (CyTOF) and have suggested that the classical nomenclature may require adjustment to accurately represent the true heterogeneity of neutrophils. Evrard et al. performed CyTOF analysis on murine BM cells and revealed the existence of three neutrophil subsets including a proliferative neutrophil progenitor group (preNeu), and non-proliferating immature and mature groups (49). PreNeu express CD117 (c-kit) but do not express markers for other leukocyte lineages. Phenotypically, preNeu were defined as Lin-c-kitintCD11b+CXCR4+ cells containing primary and secondary granules. The human counterparts of the preNeu subset were defined as being Lin-CD34-CD101-CD15+CD66b+ cells. Immature and mature neutrophils can be discriminated based on the distinct expression profiles of CXCR2 and CD101. Immature neutrophils are Lin-c-kit-CD11b+CXCR4-CXCR2-CD101- (or Lin-CD11b+CD115-Ly6GlowLy6Bint) and are mainly expressing secondary granule proteins. Mature neutrophils are CD11b+CD115-Ly6G+CXCR2+CD101+ and express gelatinase granule proteins (49). In humans, immature and mature neutrophils are defined as Lin-CD34-CD101+CD15+CD66b+CD10-CD16- and CD101+CD15+CD66b+CD10+CD16+, respectively (53, 54). Kim et al. confirmed the existence of a murine neutrophil precursor and defined these cells as Lin-c-kit+CD11b+Ly6GlowLy6BintCD115-GFI1+ cells (55). Later Zhu et al. showed that two populations of neutrophil precursor cells can be distinguished based on the expression of CD34. Early-stage and late-stage precursor cells were defined as c-kit+Gfi1lowCEBPAhiLy6Glow and c-kit+Gfi1highCEBPAlowLy6G+ cells, respectively (56).
Circulating Pool of Neutrophils in Blood
Mature neutrophils can be stored in the BM and are liberated into the circulation upon appropriate stimulation (Figure 4) (1, 7). Recent scRNA-seq experiments have exposed an additional layer of complexity by showing that neutrophils can enter the peripheral blood without going through the most mature stage first, entering the circulation as immature cells (57). The balanced action of the chemokine receptors CXCR4 and CXCR2 tightly regulates the release of neutrophils. Upregulation or downregulation of CXCR2 and CXCR4, respectively, is associated with egress from the BM (58). CXCR4 is the main receptor for CXCL12, which is expressed by the stromal cells of the BM and retains developing neutrophils within the BM (11). Enhanced activity of CXCR4 delays the release of mature neutrophils from the BM as seen in patients with WHIM (‘warts, hypogammaglobulinemia, infections, and myelokathexis’) syndrome, a rare primary immunodeficiency disorder (59). Also, the adhesion molecule vascular cell adhesion molecule (VCAM)-1, expressed by the BM epithelial cells, promotes retention of neutrophils within the BM via its interaction with the integrin very late antigen (VLA)-4 on neutrophils (11). In contrast, human CXCR2 - which recognises CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8 - stimulates the release of neutrophils from the BM. Granulocyte colony-stimulating factor (G-CSF) further promotes neutrophil mobilization by lowering CXCL12 production (60) and CXCR4 expression (61) and by increasing the amounts of mobilising signals (e.g. CXCL1) (62). The release of neutrophils outside the BM is accompanied by the release of granule proteins including MMP-9, paving the transendothelial/transcellular way through the sinusoidal endothelium (63, 64). Moreover, in human, the most potent chemokine activator of CXCR2, i.e. CXCL8 is further potentiated by N-terminal truncation by MMP-9 creating a positive feedback loop and inhibition of CXCL8 with neutralizing anti-MMP-9 antibodies inhibited CXCL8-induced mobilization of progenitor cells from bone marrow in monkeys (65, 66). Interestingly, the transit from the BM to the peripheral blood is associated with large transcriptomic and epigenetic differences (67).
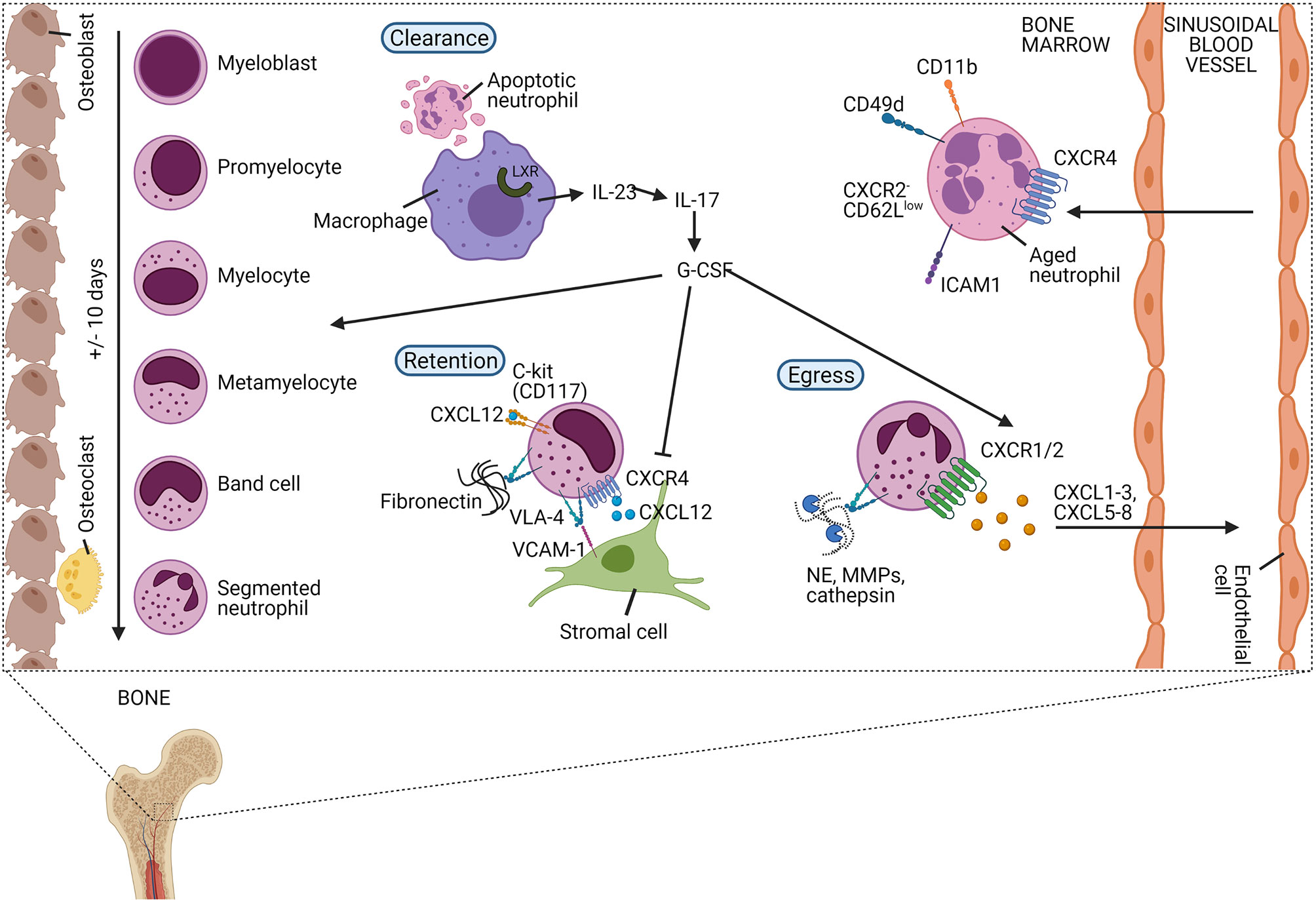
Figure 4 The release of neutrophils from the bone marrow is tightly regulated in homeostatic conditions. In the bone marrow, neutrophils differentiate from undifferentiated hematopoietic stem cells (HSCs) into mature neutrophils via different immature stages of myeloblast, promyelocyte, myelocyte, metamyelocyte, immature band cells, and mature segmented neutrophils. Neutrophils are retained in the bone marrow (BM) via the homing CXCR4 receptor or the stem cell antigen CD117 (C-kit) which both bind CXCL12, released by stromal cells. Additionally, the integrin very late antigen 4 (VLA-4) expressed on neutrophils are retaining neutrophils in the BM by binding the adhesion molecule vascular cell adhesion protein-1 (VCAM-1) or fibronectin. Granulocyte colony-stimulating factor (G-CSF) is an important growth factor that regulates the proliferation, differentiation, and egress of neutrophils in the BM. It aborts the CXCR4/CXCL12 interactions and releases neutrophilic chemoattractants including CXCL1-3 and CXCL5-8 that bind CXCR2. During the release of neutrophils from the BM, granule proteins are released degrading the fibronectin and mediating trans-endothelial transport. Aged neutrophils are characterised by the expression of diverse receptors including upregulated CXCR4, ICAM1, CD11b, CD49d, and low expression of CXCR2 and CD62L. Aged neutrophils migrate back to the BM where they are engulfed by macrophages. Upregulation of the liver X receptor (LXR) family induces the expression of interleukin-23 (IL-23) that may enhance the release of new neutrophils, balancing the homeostasis of neutrophils, via the IL-23/IL-17/G-CSF-axis.
Marginated Pools of Neutrophils in Organs
Margination refers to the prolonged transit of neutrophils through organs, which results in discrete intravascular (marginated) pools. These can be found within the spleen, liver, and BM. Lung margination may be specific only for certain species such as primates, mice, and dogs (1, 58, 68–70). During infection or inflammation, cytokine or epinephrine (adrenaline) stimulation can quickly release the marginated neutrophils into the circulation (71–73).
Extravasation of Neutrophils Into Inflamed Tissues
Effective pathogen elimination requires the presence and activation of neutrophils at the right location in the body. Neutrophils need to enter the inflamed tissue via extravasation, a process that is coordinated by selectins, integrins, and soluble mediators including proteases and chemoattractants (Figure 5). The different chemoattractants are classified into chemotactic lipids [e.g. leukotriene B4 (LTB4)], chemokines [CXCL1 to CXCL3 and CXCL5 to CXCL8 in humans and KC, macrophage inflammatory protein-2 (MIP2), and granulocyte chemotactic protein-2 (GCP-2) in mice], complement anaphylatoxins (C3a and C5a), and formyl peptides [e.g N-formylmethionyl-leucyl-phenylalanine (fMLF)]. Their specific roles in the regulation of neutrophil migration and activation were recently reviewed by Metzemaekers et al. (74–76).
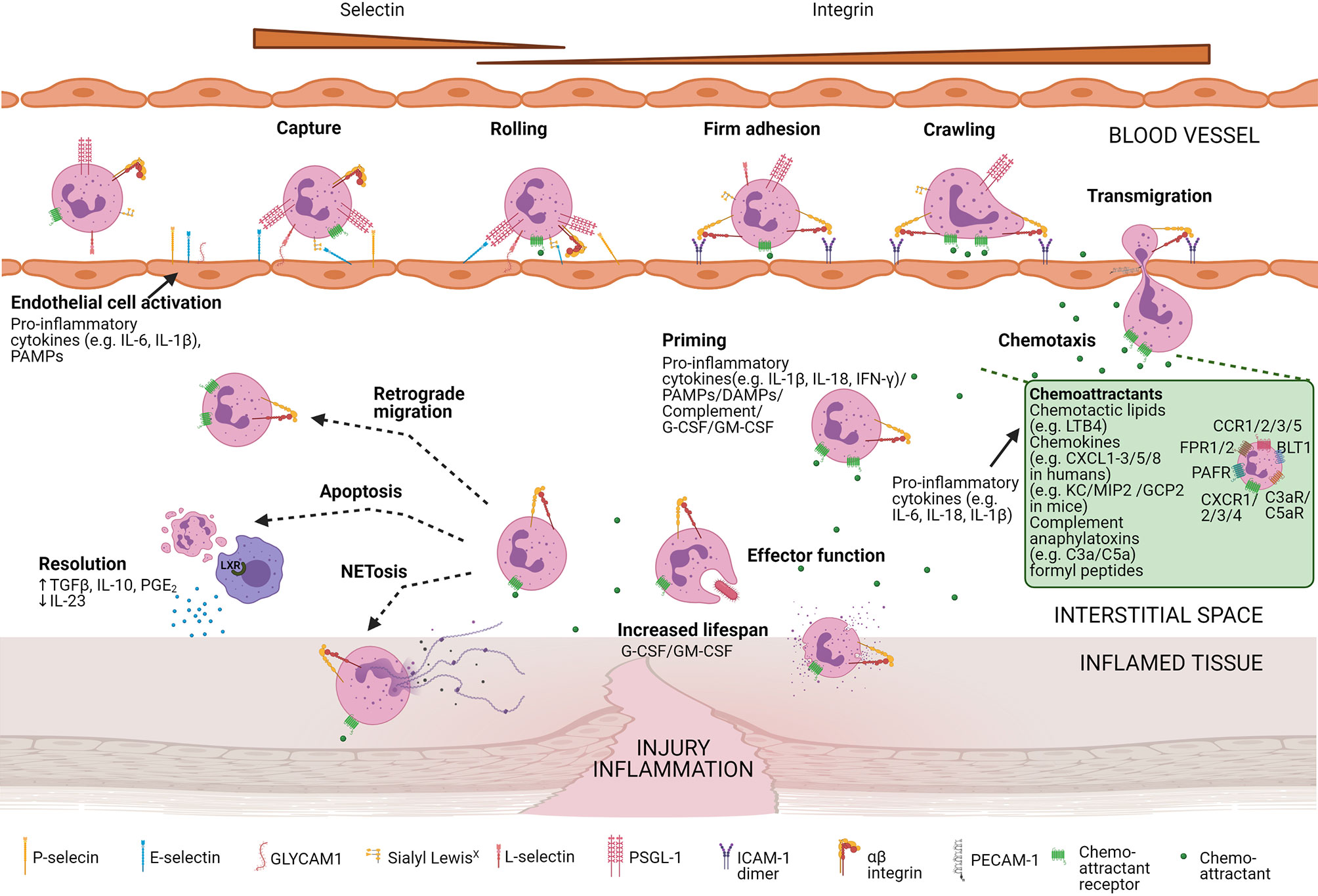
Figure 5 Neutrophil recruitment towards the site of inflammation. Neutrophil mobilisation and internalisation involve different steps including the capture of the cells, rolling, firm adhesion, crawling, and transmigration. Diverse neutrophil surface receptors are involved in this process. Pro-inflammatory cytokines or pathogen-associated molecular patterns (PAMPS) activate endothelial cells, allowing them to express adhesion molecules (e.g. P-selectins and E-selectins). These adhesion molecules bind mucins expressed on the neutrophil surface capturing and slowing the neutrophil. Slow rolling of the cells allows chemoattractants to bind their respective chemoattractant receptors. Subsequent signalling results in integrin activation and eventually firm adhesion. Next, neutrophils are crawling along with the endothelial cells and transmigrate at cell-cell junctions (paracellular migration) or by endothelial cell bodies (transcellular migration). Once migrated, neutrophils are further attracted towards the centre of inflammation by a gradient of chemoattractants. An overview of the main chemoattractants is shown in the green box. Chemoattractants are mainly released by pro-inflammatory cytokines. At the focus of inflammation, neutrophils are primed by proinflammatory cytokines, PAMPs, damage-associated molecular patterns (DAMPs), complement molecules, and/or growth factors enhancing their effector function and additionally increases their lifespan. In vivo studies in animals suggest that extravasated neutrophils can back-migrate (retrograde migration). Otherwise, neutrophils undergo NETosis or apoptosis. Apoptotic neutrophils are engulfed by tissue-resident macrophages that upregulate transcription factors of the liver X receptor (LXR) family, potentiating resolution at the site of inflammation.
Neutrophil extravasation encompasses four well-defined steps. First, pro-inflammatory cytokines (including tumour necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-17) or stimulants of bacterial origin (including LPS) induce the upregulation of P-selectins and E-selectins on endothelial cells. These adhesion molecules can bind to glycoproteins e.g. P-selectin glycoprotein ligand-1 (PSGL-1) present on the surface of neutrophils, mediating rolling of the latter along the endothelium. Next, chemoattractant-induced signalling and interaction between L-selectin (CD62L) and its ligands (e.g. GlyCAM-1) on endothelial cells can activate integrins such as lymphocyte function-associated antigen 1 (LFA-1) on neutrophils. LFA-1 binds to ICAM1 and ICAM-2 on endothelial cells and mediates firm neutrophil adhesion and arrest. Afterwards, neutrophils crawl along the endothelium and exit the blood vessel preferentially via the paracellular route. Neutrophils may also follow a transcellular path directly through the endothelial cell body without the loss of the integrity of the plasma membrane of either cell. Finally, neutrophils further migrate towards increasing concentrations of chemoattractants (1, 77–83).
Neutrophilic Functions at the Site of Inflammation
Neutrophils have diverse functions which are illustrated in Figure 6 (1, 4, 5). A full description of the different neutrophilic functions falls out of the scope of this review and has been provided elsewhere (1). Main antimicrobial functions include: ROS production, degranulation, neutrophil extracellular trap (NET) formation, phagocytosis and ectosome formation (Figure 6A). Whereas the degranulated granule proteins may tag (i.e. by the binding of cationic antimicrobial peptides such as defensins or cathelicidins) or eliminate extracellular pathogens [i.e. by neutrophil elastase (NE), MPO, or lactoferrin], they may also induce tissue damage and neutrophil migration (i.e. by MMP-8 or MMP-9) (1, 47, 78, 84, 85). The secreted granule proteins also influence the inflammatory activities of the neighbouring cells and promote cytokine, chemokine, ROS production, cell activation, or cell extravasation.
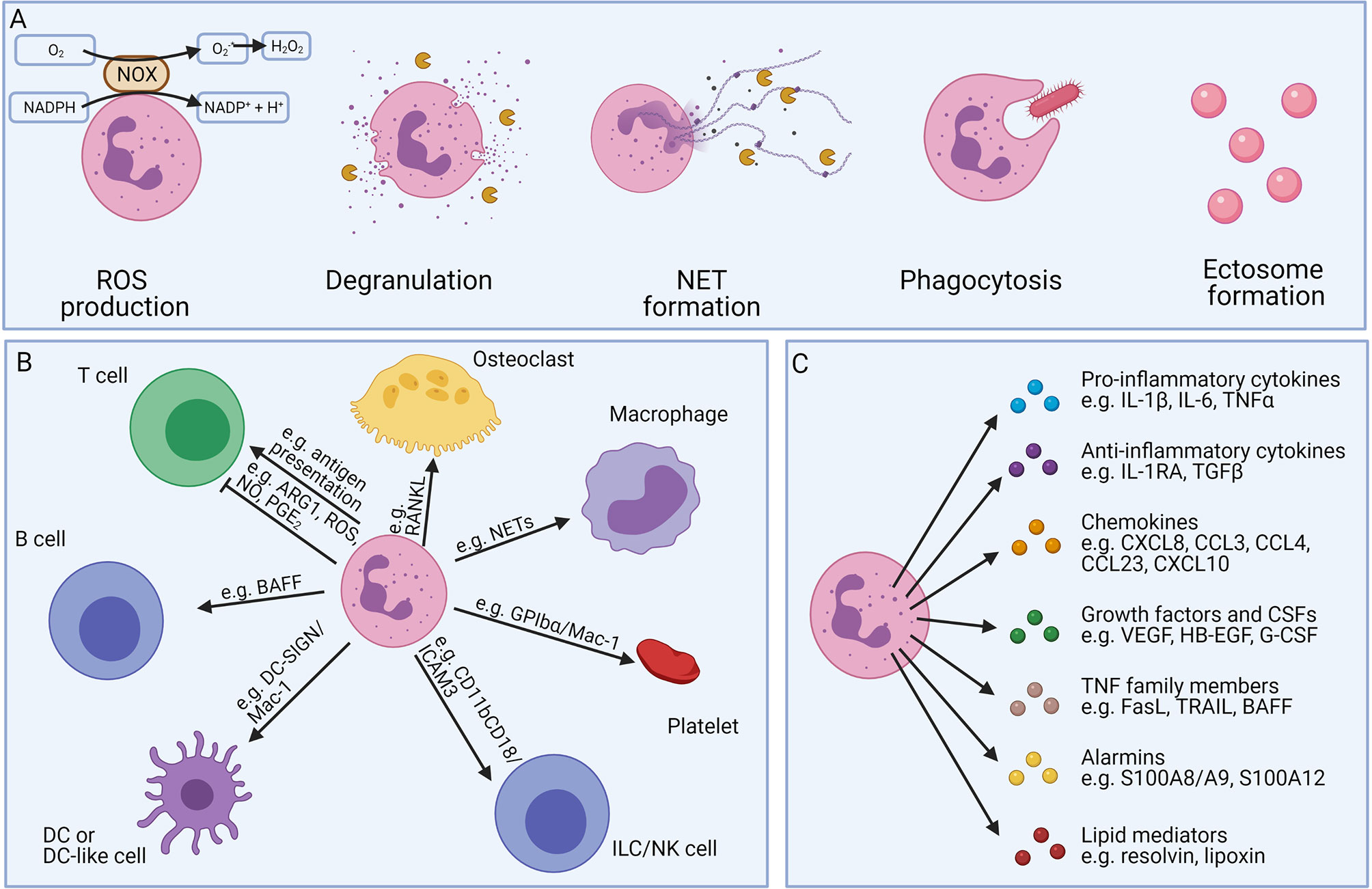
Figure 6 Overview of the different neutrophil effector functions. Neutrophils possess different defence mechanisms including both oxidative and non-oxidative mechanisms (A). Neutrophils can produce reactive oxygen species (ROS) and release soluble mediators stored in pre-made granules (degranulation). Neutrophils show a unique form of cell death, called NETosis, where neutrophils expel neutrophil extracellular traps (NETs), consisting of decondensed DNA together with histones, cytokines, and granule proteins. Furthermore, neutrophils are professional phagocytes able to engulf foreign particles for internal digestion. Neutrophils can also transform into anuclear cytoplasts (ectosomes), regulating the inflammatory microenvironment, intercellular communication, and exerting antimicrobial functions. Neutrophils interact with different cells of both the innate and adaptive immune systems (B). Neutrophils can carry antigens and act as antigen-presenting cells, activating T cells. In contrast, myeloid-derived-suppressor neutrophils can inhibit the proliferation of T cells. Neutrophils can stimulate the maturation of antibody-producing B cells and play an essential role in the generation of natural killer (NK) cells in the bone marrow. Furthermore, neutrophils may also interact with platelets, macrophages, dendritic cells (DCs), or osteoclasts. Neutrophils also release a bunch of soluble mediators (C). Although the release per individual cell is limited, neutrophils are an important source, due to their high abundance in the blood.
Next to the important antimicrobial properties, neutrophils can also directly or indirectly interact with other immune cells (including T cells, B cells, NK cells, macrophages, and DCs) and are critical in establishing a good immune response (summarised in Figure 6B). Neutrophils can secrete both pro-inflammatory cytokines (e.g. TNF-α, IL-18, and IL-1β), anti-inflammatory cytokines (i.e. IL-10, and IL-1RA), and chemokines (e.g. CCL2, CXCL8, CXCL9, CXCL10, and CCL20), further recruiting new immune cells to the site of inflammation (86–91). Despite the relatively low amounts of cytokines/chemokines produced by neutrophils and their limited capacity for de novo protein synthesis, neutrophils and their activation products can play significant roles during inflammation since they are usually overwhelming in terms of absolute cell numbers (92). The neutrophil-derived proteases may additionally modulate the signaling network via their cytokine and chemokine processing capacity (93–95). Neutrophils also produce growth factors, alarmins, and angiogenic factors (G-CSF, S100 proteins, and VEGF) via which they may influence different biological processes (86, 96, 97) or may induce immunoglobulin class switching and somatic hypermutations in B cells by the secretion of B-cell activating factor (BAFF), a proliferation-inducing ligand (APRIL), also known as TNF ligand superfamily member 13 (TNFSF13), and IL-21 (98–101). Neutrophils can also regulate the inflammatory outcome by changing the composition of the secreted lipids (Figure 6C) (4).
At the site of inflammation, neutrophils can be primed by cytokines (IL-1β, IL-6 or TNF-α), pathogen-associated molecular patterns (PAMPs; e.g. LPS), damage-associated molecular patterns (DAMPs; e.g. ATP), or complement-opsonized particles via their vast repertoire of cytokine receptors and pathogen recognition receptors (PRRs) (102). Primed neutrophils are phenotypically defined by increased CD16 or CD11b/CD18 expression levels and reduced expression of CD62L. In comparison with quiescent cells, primed neutrophils display a more aggressive action upon subsequent activation with a second inflammatory stimulus, illustrated by enhanced ROS production, degranulation, phagocytosis, and an increased tendency to release NETs (103).
Apoptosis and Neutrophil Clearance
Under homeostatic conditions, senescent ‘aged’ neutrophils upregulate CXCR4 and CD11b while downregulating CD62L (104, 105). Aged neutrophils transmigrate into the BM where they are cleared by the resident stromal macrophages. This results in the induction of transcription factors belonging to the liver X receptor (LXR) family. Macrophages and LXRs are essential components for the modulation of the hematopoietic niche and may cause G-CSF production and downregulation of CXCL12, which subsequently result in an increased neutrophil mobilisation (106). Using similar mechanisms, hypocellularity (e.g. during antibody depletion), can also be sensed in the BM. In addition, circadian rhythm and food intake via microbiota-derived signals also regulate the number, maturation, and gene expression program of neutrophils. A full description of the intrinsic clockworks or diurnal rhythm variations in systemic and local factors falls out of the scope of this review [reviewed in (107–110)].
After fulfilling their effector functions at the site of inflammation, neutrophils may undergo suicidal NET formation, necrosis, or apoptosis. Apoptotic neutrophils can be phagocytosed by resident macrophages and subsequently upregulate LXR (111, 112). This induces a reduced IL-23 production and subsequent reduced IL-17 and G-CSF that terminates the inflammatory recruitment of additional neutrophils, tempering inflammation and restoring homeostasis. Furthermore, it stimulates the secretion of the anti-inflammatory cytokines TGF-β and IL-10, which further decrease the neutrophil chemo-attraction and activation (6).
Emergency Granulopoiesis in sJIA
Emergency Granulopoiesis and Extramedullary Myelopoiesis
During excessive inflammation, neutrophil homeostasis is disturbed. Neutrophils are massively attracted to the site of inflammation, their clearance is decreased, and their lifespan is increased. Cytokine stimulation can quickly release neutrophils from the marginated pool and the high demand for new neutrophils results in a massive generation of new neutrophils (outside the BM) described as emergency granulopoiesis and extramedullary myelopoiesis respectively (Figure 1, red arrows).
Emergency granulopoiesis involves a series of conserved cascading events and is especially well documented during infection (113, 114). The different steps involved are summarised in Figure 7. Emergency granulopoiesis is characterised by an increased release of immature neutrophils, such as myelocytes, metamyelocytes, and band cells into the circulation, known as ‘the left shift’ (115). Clinically, the left shift is defined by leucocytosis with the appearance of immature neutrophil precursor cells in the peripheral blood, which are normally only present in the BM. In sepsis, the presence of immature neutrophils in the circulating blood is often used as a clinical indicator (116, 117). In humans, mature and immature neutrophils can be distinguished based on the CD10 expression which is restricted to mature neutrophils (54). Furthermore, based on expression levels of CD16 and CD62L (L-selectin) neutrophils can be subdivided into mature, immature, and hyper-mature neutrophils. In non-inflammatory conditions, nearly all circulating neutrophils have a mature phenotype and are CD16highCD62Lhigh cells. In inflammatory settings, an increased number of immature neutrophils (CD16dimCD62Lhigh) and hypersegmented neutrophils (CD16highCD62Ldim) are found (118–120). Recently, a single-cell sequencing experiment was performed in a bacterial infection-induced mouse model. In this study, researchers showed that emergency granulopoiesis is associated with an augmented proliferation of the early-stage neutrophil progenitor cells in the BM and accelerated post-mitotic maturation of the neutrophils. Interestingly, the overall neutrophil differentiation in the BM remains intact but a substantial difference in the transition between the subpopulations was reported (57).
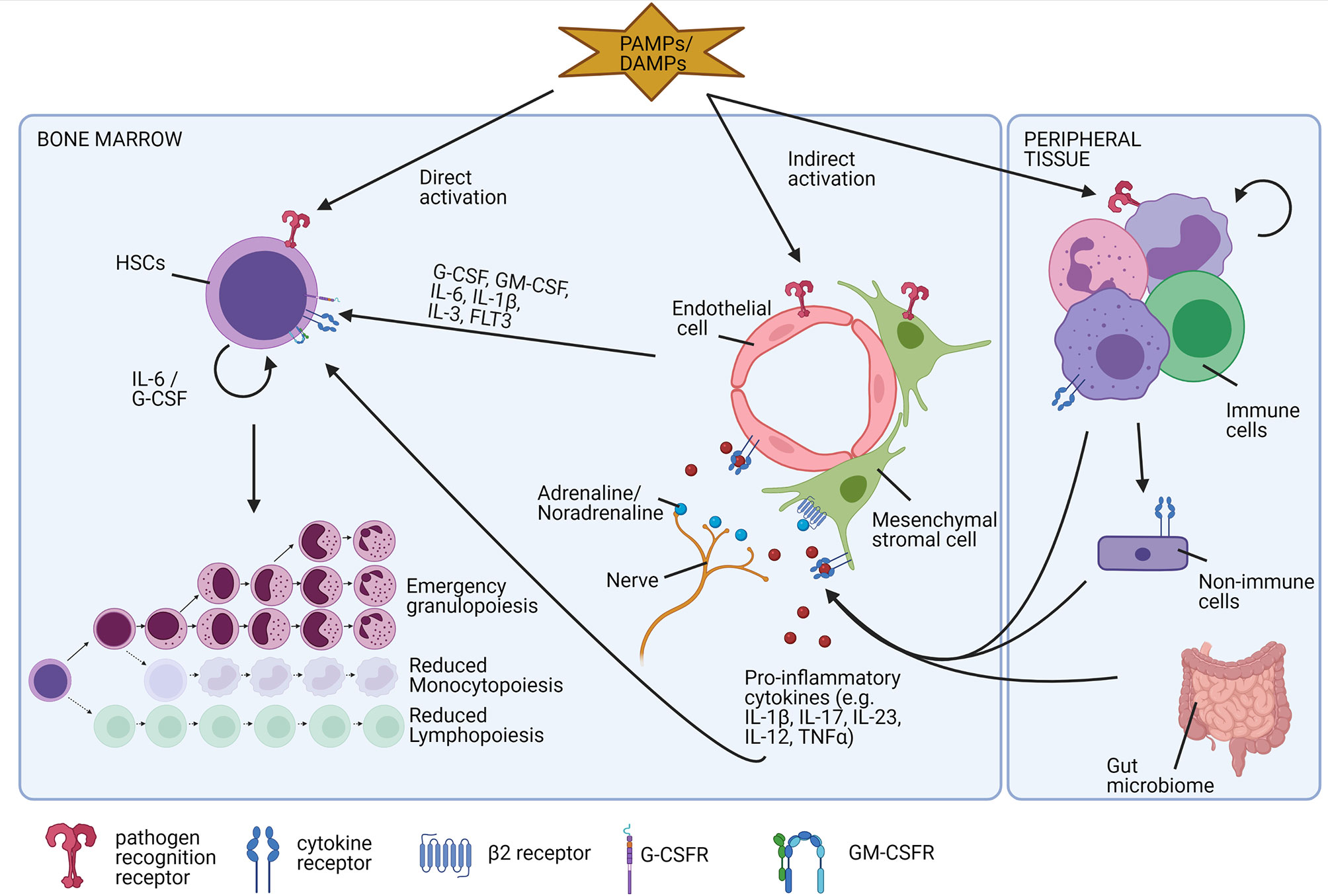
Figure 7 Factors involved in the induction of emergency granulopoiesis. During excessive inflammation, there is a high need for new neutrophils (emergency granulopoiesis). Different direct and indirect mechanisms are involved in the differentiation and release of new neutrophils. Damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) bind diverse pathogen recognition receptors, expressed on diverse cell types. In hematopoietic stem cells (HSCs), binding directly stimulates the release of interleukin-6 (IL-6) and granulocyte colony-stimulating factor (G-CSF), inducing their proliferation and differentiation into neutrophils. Indirectly, the molecular pattern molecules bind endothelial cells or stromal cells, providing an important source of pro-inflammatory molecules and growth factors. Additionally, these endothelial cells or mesenchymal cells are stimulated by pro-inflammatory cytokines released by immune cells or non-immune cells of the periphery that encounter invading pathogens or cell damage. The neuronal release of adrenalin or noradrenalin additionally stimulates the release of new neutrophils. Finally, also food intake and the gut microbiome tightly regulate neutrophil homeostasis.
Today, it remains challenging to accurately discriminate between steady-state and emergency haematopoiesis. Studies aiming to dismantle the mechanisms involved in emergency granulopoiesis have shown that granulopoiesis can be induced by cytokines in the absence of C/EBPα and suggested alternative pathways under emergency conditions (Figure 8) (121). It was found that especially C/EBPβ plays a crucial role in the stress-induced haematopoiesis, which was hampered in C/EBPβ KO mice (16, 121–127). Whereas both C/EBPα and C/EBPβ share downstream genes involved in the granulocyte differentiation (128), a less pronounced cell cycle inhibition was linked to C/EBPβ as compared to C/EBPα (37, 121, 129–131). The importance of C/EBPβ in stress-induced granulopoiesis has been confirmed in diverse mouse models and a zebrafish model (132–134). Interestingly, in chronic myeloid leukaemia, the breakpoint cluster region-Abelson murine leukaemia virus (BCR-ABL) fusion protein may drive a myeloid expansion by activating the emergency granulopoiesis in a C/EBPβ-dependent way (123, 124).
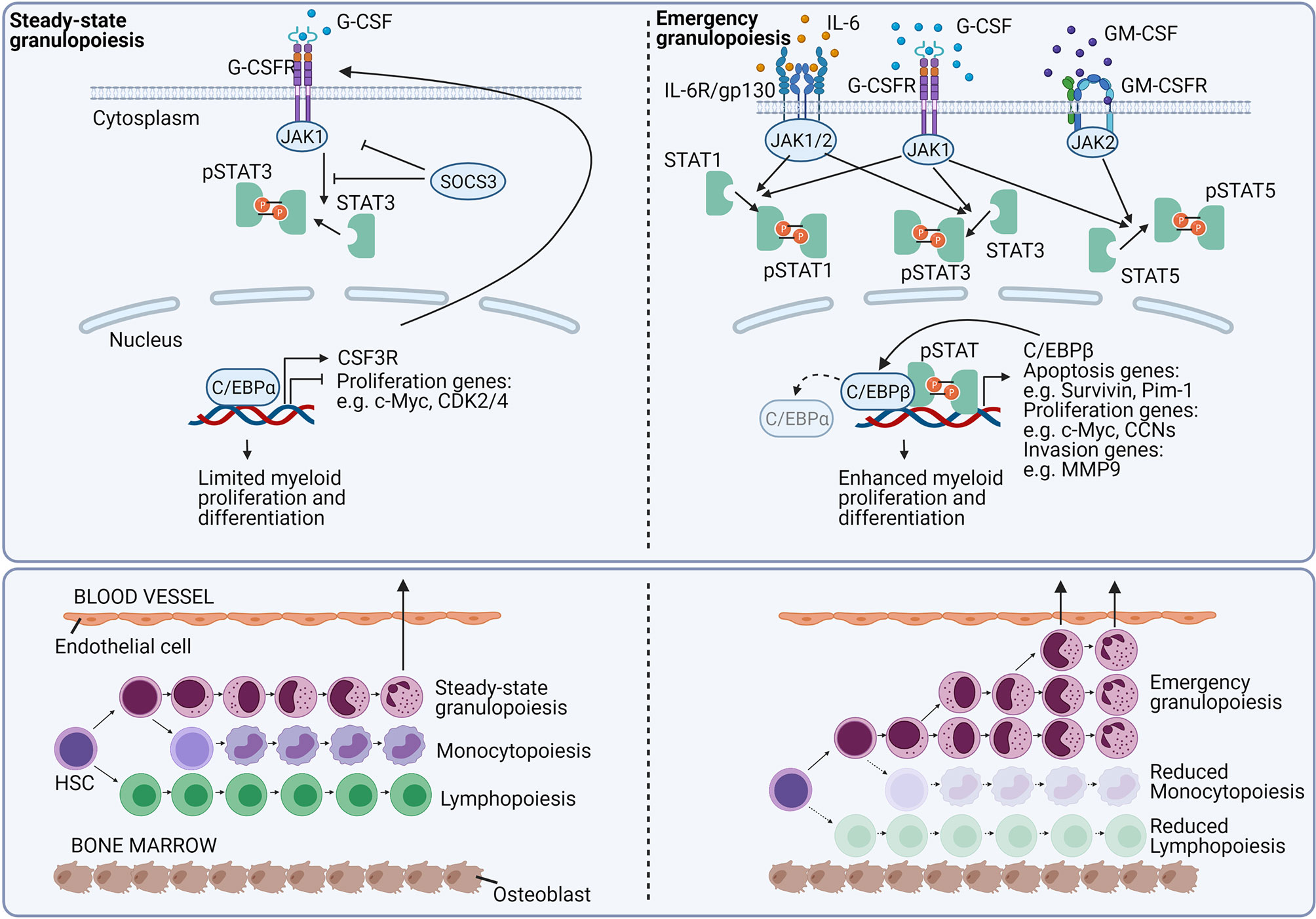
Figure 8 Growth factor signaling regulates emergency granulopoiesis. In homeostatic conditions, the granulocyte colony-stimulating factor (G-CSF) is the main growth factor that regulates neutrophil development. G-CSF signals via the G-CSF receptor (G-CSFR) (encoded by the CSF3R gene), and signals via the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. Via unknown mechanisms, G-CSFR signaling induces the CCAAT-enhancer-binding protein-α (C/EBPα). C/EBPα is the major transcription factor involved in myelopoiesis. The transcription factor enhances the expression CSF3R, whereas it inhibits genes required for the cell cycle, eventually causing a limited myeloid proliferation and differentiation. During excessive inflammation, neutrophils massively migrate to the site of inflammation. To counterbalance neutrophil depletion and to provide newly needed neutrophils, emergency granulopoiesis is initiated. Emergency granulopoiesis is characterised by a large-scale de novo generation of new neutrophils from neutrophil progenitor cells. Interleukin-6 (IL-6), G-CSF, and granulocyte-macrophage colony-stimulating factor (GM-CSF) are mainly stimulating the proliferation and differentiation of new neutrophils. IL-6 binds the IL-6 receptor (IL-6R)/gp130 and signals in a JAK-STAT1 or STAT3-dependent way. Excessive G-CSFR signalling induces both STAT1, STAT3, and STAT5 phosphorylation and signalling, whereas GM-CSF receptor (GM-CSFR) signalling mainly tempts STAT5 activation. Upon activation, the pSTAT molecules are translocated to the nucleus where they directly stimulate genes involved in the regulation of apoptosis, proliferation, and cellular translocation. Additionally, pSTAT signalling stimulates the expression of the transcription factor C/EBPβ. C/EBPβ replaces C/EBPα, and releases the brake on proliferative genes, cranking myelopoiesis. During emergency granulopoiesis, not only mature neutrophils are leaving the BM, but also more immature neutrophils are being released. This process is called “left shift”.
During extramedullary myelopoiesis neutrophils are formed outside the BM. The extramedullary myelopoiesis mainly takes place in spleen, and liver and, more controversially, also in lymph nodes, lungs, and kidneys (16, 135–139). Under acute or chronic stress conditions such as infection, these HSPC can seed in the extramedullary tissues (138). In the murine spleen, the HSPCs were localised close to transcription factor 21 (Tcf21)-positive stromal cells that can secrete niche factors such as CXCL12 and stem cell factor to support the splenic extramedullary haematopoiesis EMH (140). Although the BM niche and splenic stroma are quite different, neutrophil development in the spleen during extramedullary haematopoiesis is believed to follow the same hierarchical developmental order as described in the mouse BM. In this way, the spleen provides a unique reservoir able to supply additional myeloid cells during the immune challenge (49, 141, 142).
Emerging evidence from cancer studies suggests that neutrophils generated from the BM are functionally different from those derived from extramedullary sites such as the spleen. Driven by the cancer microenvironment, HSPCs can generate myeloid-derived suppressor cells (MDSC) in the spleen that can suppress the activation and proliferation of T cells (141, 143, 144). Interestingly, C/EBPβ can regulate the expression of enzymes such as arginase and inducible nitric oxide synthase both of which are required for the lymphocyte inhibitory activities of the MDSC (56) and the absence of C/EBPβ could reduce the tumour metastasis (145, 146). Pre-existing differences in the chromatin landscape between the different neutrophil maturational stages may contribute to observed heterogeneity since the same environmental trigger may induce a different biological output (111).
The term ‘reactive granulopoiesis’ has been proposed in settings where emergency granulopoiesis is induced by a non-infectious trigger such as described in active sJIA patients. sJIA patients show an increased number of circulating (immature) neutrophils which is in line with the peripheral expansion of immature CD34+CD33+ myelomonocytic precursors (147, 148). In addition, increased levels of neutrophil-derived mediators such as S100 proteins, MMP-8, MMP-9, elastase, and proteins involved in adhesion and chemotaxis of neutrophils (e.g. soluble E-selectin and soluble ICAM-1) were measured in the plasma of patients with sJIA. Furthermore, a positive correlation was found between increased numbers of neutrophils and levels of the inflammatory mediators C-reactive protein (CRP), ferritin, S100A8/A8, S100A12, MMP-8, and soluble E-selectin (149). Since neutrophils are thought to drive the pathogenesis of sJIA, in the next paragraph, we describe how the different neutrophil functions including the different neutrophil subsets may contribute to the disease pathogenesis of sJIA.
How Neutrophilia May Fuel the Pathogenesis of sJIA
sJIA is a childhood autoinflammatory disease characterised by expansion of neutrophils. The disease can also occur in adults, where it is called Adult-onset Still’s disease (AOSD) (150). Apart from an important neutrophilia, patients are diagnosed by the presence of arthritis in one or more joints with or preceded by a quotidian fever of at least 2 weeks and accompanied by an erythematous rash, and enlargement of the lymph nodes (lymphadenopathy), liver and/or spleen (hepatosplenomegaly) or serositis (151). Patients also present with fatigue, abdominal pain, and weight loss (148, 152–154). In addition to neutrophilia, laboratory findings include (normo- or microcytic) anaemia, thrombocytosis, and a high erythrocyte sedimentation rate (ESR). Plasma levels of inflammatory cytokines (IL-6, IL-18), acute phase proteins S100A8/A9, and S100A12, and CRP are highly increased. In addition, the level of ferritin and D-dimers and liver enzymes such as aspartate transaminase (AST), alanine transaminase (ALT), and lactate dehydrogenase (LDH) are often elevated as well (148, 152, 154).
The exact aetiology of sJIA remains enigmatic. sJIA is not an infectious disease since, up till now, the condition has not been consistently associated with any pathogen. Given a seasonal variation in some studies, it has been postulated that infectious agents may trigger an excessive immune reaction to a relatively harmless trigger in genetically susceptible children (148, 152, 155). Clinically, sJIA follows a biphasic clinical course, in which the innate immune system is mainly involved at disease initiation (febrile stage) with excessive activation of neutrophils, monocytes, natural killer (NK) cells, and γδ T cells (156). The constitutive activation of the innate immune system supports the notion that describes sJIA as an autoinflammatory disorder. Several immunological genetic polymorphisms have been demonstrated to be associated with sJIA, defining sJIA as a multigenic and multifactorial autoinflammatory disease (157–169). In contrast to monogenic autoinflammatory diseases, also the adaptive immune system is involved in the pathogenesis of sJIA and contributes to the development of arthritis at a later stage (arthritic stage). Evidence for involvement of adaptive immunity in the pathogenesis of the disease is further provided by the fact that expression of the human leukocyte antigen (HLA) variant HLA-DRB1*11 places paediatric individuals at risk for developing sJIA.
Neutrophils from sJIA patients show a primed phenotype, characterised by an increased intracellular ex vivo ROS production upon formyl peptide stimulation and by an enhanced secretory vesicle degranulation (170). The increased degranulation was demonstrated by the increased surface expression of the complement receptor CD35, upregulation of the high-affinity Fcγ receptor CD64, and by the enhanced release of S100A8/A9 upon stimulation with phorbol 12-myristate 13-acetate (PMA) (149). Neutrophils from patients with AOSD also show upregulation of CD64 (170). It remains unknown if neutrophil-derived ectosomes are also altered in the sJIA pathogenesis. NETs may have a dual role in autoimmune and autoinflammatory diseases. As a disease tempering role, they catch pro-inflammatory cytokines and proteolytically degrade them to resolve the inflammation (171). In contrast, NETs are also highly immunogenic and were linked to the development of several autoimmune diseases including RA and systemic lupus erythematosus (SLE) (172–180). In sJIA patients, NETosis was never directly studied ex vivo. Nevertheless, increased serum histone levels in active sJIA patients compared to inactive patients or healthy controls (HCs) were described, arguing for enhanced NETosis. In addition, the serum histone levels were correlating to the sJIA disease activity (181). The increased levels of high mobility group box 1 protein (HMGB1) in patients with sJIA (182) is linked to enhanced NETosis since Garcia-Romo and colleagues have demonstrated in SLE that HMGB1 is released during NETosis, forming a positive feedback loop (183).
In sJIA, diverse clinical features in the febrile and the arthritic stage may be linked to neutrophils. At the initial stages, neutrophils are an important source of pro-inflammatory cytokines, chemokines, molecular mediators, proteases, and growth factors as blood neutrophil transcriptome analysis showed an inflammatory gene expression profile. Upregulated genes included neutrophil granule proteins, members of the IL-1 cytokine family, components of inflammasomes, the high-affinity IgG receptor CD64, CXCL8, and genes involved in the NF-κB pathway. Interestingly, the gene expression profile was partially overlapping with the transcriptome of sepsis (149, 184, 185). Additionally, using the sJIA mouse model we demonstrated that neutrophils are the main source of IL-1β (unpublished results from our own group). Worthy of note is the fact that neutrophils have a relatively high steady-state expression of pro-IL-1β and do not necessarily need a priming signal. Interestingly, patients displaying high numbers of neutrophils or neutrophil-associated genes appear to benefit from treatment with IL-1-targeting drugs such as anakinra and canakinumab (149, 168, 186). In addition, neutrophils provide an important source of alarmins or DAMPs, further amplifying the activation of the innate immune system. In sJIA, there is a growing interest in these molecules, in particular in the family of S100 proteins. S100 proteins may activate innate immune cells, predominantly monocytes and macrophages, upon binding the RAGE receptor or TLR4 by enhancing the secretion of pro-inflammatory molecules (187). High concentrations of S100 proteins (both S100A8/A9 and S100A12) are measured in patients with sJIA and therefore these molecules were proposed to be biomarkers (188–195). The release of IL-1 and other inflammatory cytokines by total white blood cells (WBCs) or monocytes from sJIA patients was reduced upon depleting serum S100A8/A9 or by preventing the S100A12 complex formation (191, 196, 197).
In the arthritic stage of sJIA, neutrophils may drive the development of arthritis fitting the remnant epitopes generate autoimmune (REGA) model that states that cells and molecules of the innate immune system (including neutrophils) can start the autoimmune reaction by cytokine-regulated proteolysis yielding remnant epitopes (2). Indeed, activated neutrophils can destroy the cartilage by e.g. releasing granule proteins or ROS production (198, 199). This was mainly investigated and reported in rheumatoid arthritis (RA) (172, 200, 201). Additionally, neutrophils express a functionally active membrane-bound RANKL, the ligand for receptor activator of NF-kB (RANK) and so can activate osteoclastogenesis (202). In the joints of JIA patients, activated neutrophils were abundantly present (203–205) and the presence of S100A12 in synovial fluids from JIA patients (189) provides evidence for their involvement in disease pathogenesis.
Neutrophil Subsets in sJIA Patients
In sJIA patients, an increased number of immature CD16dim neutrophils has been reported. The increased percentage of immature neutrophils in the peripheral blood cell count (banded neutrophils and granulocyte precursors) was confirmed microscopically (149). Also, patients with sJIA had a higher proportion of CD62Llow neutrophils compared to healthy controls (149, 184). The shedding of CD62L, mediated by membrane-proximal cleavage, is indicative of priming (as described above) (103, 206–208). Whereas one study failed to show an increased number of hypersegmented neutrophils, another study showed, by imaging cytometry, that patients with systemically active disease have increased numbers of hypersegmented neutrophils (149, 184). We recently uncovered neutrophil-DC hybrid cells (expressing both neutrophil and DC markers) in the synovial fluid from patients with JIA (205) that may serve as antigen-presenting cells (209, 210), eventually contributing to the arthritic phenotype. Remarkably, neutrophil protease genes (including MMP-8, and MMP-9) could also be found in PBMC microarray datasets of sJIA patients and might reflect the presence of low-density granulocytes in the PBMC fraction, which was confirmed by flow cytometry analysis (185). Remark that also in AOSD, increased levels of low-density granulocytes were reported, correlating with disease activity (211). Single-cell sequencing on splenic neutrophils derived from the sJIA-like mouse model recently showed the emergence of MDSCs. From this, it is tempting to speculate that neutrophils, next to their pro-inflammatory properties, also may have some disease-tempering effects in sJIA (Malengier-Devlies et al., unpublished results).
Cytokines That Link Neutrophils to sJIA Pathogenesis
Diverse cytokines and growth factors play an important role in the pathogenesis of sJIA and all of them regulate important aspects of neutrophil homeostasis (16, 212–216). In a next part of this review, we overview the different neutrophil-regulating cytokines and growth factors in sJIA (including G-CSF, GM-CSF, IL-17, IL-1β, IL-6, IL-18, and IFN-γ). Considering the current beneficial effects of agents blocking IL-1- and IL-6 in sJIA (217–228), a better understanding of all neutrophil-regulating cytokines and growth factors may open new avenues for therapeutic intervention.
G-CSF
G-CSF is the main haematopoietic growth factor required for the proliferation and differentiation of haematopoietic precursor cells into neutrophils (229–234). G-CSF regulates the commitment of progenitor cells to the myeloid lineage (235), induces the proliferation of granulocytic precursor cells (236), reduces the transit time through the granulocytic compartment (236), and controls the viability of the BM neutrophil pool (233, 234, 237). Furthermore, it induces the release of mature neutrophils from the BM into the blood by the internalisation and consequent downregulation of CXCR4 (238, 239) or by downregulation of CXCL12 in the BM (Figure 4) (240). In contrast, G-CSF can also impede the CXCR2-induced neutrophil mobilisation by negatively regulating CXCR2-mediated intracellular signalling which under specific bacterial infections, functions as a negative regulator of neutrophil mobilisation (241). Both in vitro and in vivo, G-CSF acts on mature neutrophils and may enhance ROS production, adherence, phagocytosis, killing, antibody-dependent cellular cytotoxicity (ADCC), and may induce phenotypic alterations such as increased expression of CD11b (242). G-CSF also affects the expression of the pro-survival protein survivin which increases the lifespan of mature neutrophils (243, 244).
G-CSF signals via the homodimeric G-CSF receptor (G-CSFR) (245) in a manner that depends on a Janus kinase (JAK)/signal transducer and activator of transcription (STAT). G-CSF can stimulate three members of the STAT family (STAT 1, 3, and 5) (246–250). However, the role of STAT1 and STAT5 in the granulopoiesis is limited (251–256) and myeloid differentiation is mainly induced by STAT3 (257). In steady-state conditions, the G-CSFR signalling is controlled in a suppressor of cytokine signalling 3 (SOCS3)-dependent way (258) and STAT3-deficient mice are marked by peripheral neutrophilia (259–262). In steady-state granulopoiesis, the expression of G-CSFR is regulated by C/EBPα. C/EBPα drives the expression of many genes that encode proteins required for myeloid progenitor proliferation and granulocyte differentiation. Besides, it restricts excessive proliferation of neutrophil precursors by inhibiting the expression of genes required for cell cycle progressions e.g. the genes encoding MYC, cyclin-dependent kinase 2 (CDK2), or CDK4 (16, 263). During emergency granulopoiesis, when G-CSF levels are markedly increased, STAT3 is directly stimulating the expression of MYC and C/EBPβ, which in turn further stimulates MYC transcription by replacing C/EBPα at the promoter region (Figure 8) (16, 133).
G-CSFR-deficient mice have lower amounts of immature granulocytic precursors and neutrophils in the BM, show a 70-80% reduction of circulating neutrophils (due to a defective release), and are defective in certain mature neutrophil cell functions (233, 234, 264). Similarly, humans expressing dominant-negative receptor mutations in G-CSFR are neutropenic (265, 266). Additionally, mutations in the haematopoietic cell-specific Lyn substrate 1-associated protein X1 (HAX1), which contributes to the G-CSFR signalling pathway cause severe neutropenia in humans (267, 268). Interestingly, emergency granulopoiesis, however, can occur in a G-CSF-independent way and G-CSFR-deficient mice can still mount a granulopoietic response in a sterile model of peritonitis or during a C. albicans infection. In contrast, G-CSFR KO mice showed a more severe disease during infection with L. monocytogenes (234, 269–272).
The exogenous administration of G-CSF mimics the physiological responses that are observed during emergency granulopoiesis and increases the neutrophil count in peripheral blood (114, 273). Upon G-CSF treatment, also HSCs can be mobilised from the BM into the blood (274, 275). Currently, G-CSF is used in clinics to treat neutropenic individuals (276–278) or to mobilise hematopoietic progenitors for transplantation (230, 279).
Many tissues including endothelial cells, macrophages, epithelial cells, fibroblasts, and BM stromal cells produce G-CSF when stimulated with inflammatory stimuli such as LPS, IL-1β, or TNF-α (280–283). IL-17 and IL-23 have been implicated as major upstream regulators of G-CSF (discussed below) and play a crucial role in the pathogenesis of sJIA (230, 281, 284). G-CSF levels are increased in sJIA patients compared to HCs, whereas in other JIA subtypes, G-CSF was demonstrated to be a major regulator of the neutrophil gene expression signature (168, 185). In line with the pathological role of neutrophils in sJIA, the highest levels of G-CSF were found in patients with an incomplete response or nonresponse to anakinra (168).
In addition to its neutrophil regulatory properties, G-CSF induces the release of prostaglandin E2 and induces fever, one of the hallmark clinical features in sJIA (285). Furthermore, G-CSF can stimulate the peripheral sympathetic nervous system to release catecholamines (84) which may reduce the number of osteoblasts and the production of CXCL12 (286, 287). Interestingly, G-CSF is a strong inhibitor of the NK cell function, altering the receptor expression profiles and reducing the cytotoxic and cytokine producing capacity of these cells (288, 289). It is important to note that NK cells are an important group of innate immune cells involved in the pathogenesis of sJIA. The role of NK cells in sJIA has recently been reviewed by Vandenhaute et al. (290). The mutual role of both NK cells and G-CSF in sJIA requires further investigation. Using a sJIA-like mouse model, we recently showed that G-CSF regulates the development of arthritis. Additionally, we demonstrated that G-CSF stimulates extramedullary myelopoiesis in the spleen. Neutrophils followed a similar differentiation and maturation path as described in the BM, which was found to be CEBP/β-driven (Malengier-Devlies et al., manuscript under revision).
GM-CSF
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine secreted by a variety of cells. The major sources of GM-CSF include activated T and B cells, monocytes/macrophages, endothelial cells, fibroblasts but also neutrophils, eosinophils, epithelial cells, mesothelial cells, chondrocytes, Paneth cells, and tumour cells may secrete GM-CSF (291–293). In T cells, the release of GM-CSF is triggered by IL-1β and IL-23 or IL-1β and IL-12 in mice and humans respectively (294–296). In fibroblasts, endothelial cells, chondrocytes, and smooth muscle cells, TNF-α and IL-1 are the main inducers of GM-CSF production whereas in macrophages/monocytes GM-CSF is predominantly released upon TLR-stimulation (291). GM-CSF can form a positive feedback loop by activating macrophages and DCs to produce IL-23, IL-1β, and IL-6 activating Th17 and Th1 cells that in turn express GM-CSF (297). The production can be inhibited by IFN-γ (298), IL-4 (299), IL-10 (300), and glucocorticoids (301).
GM-CSF is required for the in vivo development of neutrophils, monocytes, and macrophages from BM precursor cells and plays a crucial role in the maintenance of the innate immune homeostasis (302–305). Depending on the dose, GM-CSF can have different effects on myeloid cell survival, proliferation, or differentiation (243, 306–308). GM-CSF also upregulates the antimicrobial function of mature neutrophils and enhances ROS production, adherence, killing, phagocytosis, and antibody-dependent cellular cytotoxicity both in vitro and in vivo (242, 309). Furthermore, GM-CSF phenotypically alters neutrophils and can upregulate the expression of the integrin CD11b, which facilitates adhesion and tissue entry (242, 310). Following chemo- or radiotherapy, exogenous GM-CSF can be used to restore the myeloid populations (311). In addition to its effect on neutrophils, GM-CSF stimulates the activities of macrophages, DCs, and B cells [reviewed in (312)].
GM-CSF signals via the GM-CSF receptor (GM-CSFR) that is composed of a low-affinity α chain and a high-affinity β chain. The β chain is shared with IL-3 and IL-5 receptors (307). GM-CSFR is expressed on myeloid cells and some non-hematopoietic cells, but not on T cells (313). Four main signalling pathways can be triggered by G-CSFR (314–316). The main pathway involves JAK2/STAT5 signalling and facilitates the activation of genes such as pim-1, CIS, and cyclin to induce cell myeloid differentiation and proliferation (317–319). Next, both the phosphatidylinositol-3-kinase (PI3K) and JAK/STAT-Bcl-2 signalling pathway are involved in cell survival (320). Eventually, the ERK1/2 and NK-κB pathways mediate cell differentiation and inflammation (321, 322).
Whereas GM-CSF-deficient mice have an impaired reproductive capacity and develop pulmonary alveolar proteinosis (PAP), GM-CSF-deficient mice have a normal basal granulopoiesis and show no alterations in peripheral blood counts (271, 323, 324). This indicates that other growth factors aside from GM-CSF have a redundant role in myeloid cell development and differentiation under steady-state conditions. In emergency granulopoiesis, GM-CSF plays an important role and GM-CSF-deficient mice fail to control infections with L. monocytogenes or M. avium (270, 325). In contrast, mice lacking the three myeloid colony-stimulating factors [G-CSF, GM-CSF, and macrophage colony-stimulating factor (M-CSF)] still mount an inflammatory response in a sterile model of peritonitis (272). Interestingly, GM-CSF (but also IL-3 and IL-6) can restore the lack of G-CSFR expression in C/EBPα-deficient mice and can initiate a C/EBPα-independent granulopoiesis (326–328). In vitro studies suggested C/EBPβ as the driving transcription factor in this process, since C/EBPβ-deficient hematopoietic cells had impaired responsiveness to GM-CSF (132).
GM-CSF has a pathological role in Th17-driven autoimmune diseases such as multiple sclerosis (MS) and RA (294, 297, 329). Consequently, the ablation of GM-CSF signalling could suppress the disease in models of arthritis, multiple sclerosis (MS), and lung disease (308, 330–332). In patients with JIA, the frequency of GM-CSF producing T helper cells was significantly increased in the synovial fluid and correlated with an increased ESR (333, 334). sJIA patients show increased plasma levels of GM-CSF as compared to healthy controls and a significantly decreased level was observed in the anakinra responder group (80). In the sJIA-like mouse model, depleting GM-CSF however had no effect on the observed disease symptoms (unpublished results from our laboratory).
IL-17
IL-17 is a pro-inflammatory cytokine that is mainly secreted by Th17 cells. Th17 cells are induced from naïve T cells in the presence of both TGF-β and IL-6 (in mice) or IL-1β (in humans). The pro-inflammatory cytokines TNF-α and IL-1β can synergistically increase IL-6 production, further contributing to Th17 cell differentiation. Following activation, RORγt is induced, which promotes the expression of IL-17 and the IL-23 receptor (IL-23R). Subsequent IL-23-signalling further increases the RORγt and IL-17 expression in a STAT3-dependent way (335). Although IL-17 was first described as a T cell-secreted cytokine, it can also be produced by innate immune cells such as γδ T cells (336). Since these cells do not require induction of the IL-23R, these cells may induce a fast IL-17 response to IL-1β or IL-23 without any T cell receptor engagement (337). Note that together with IL-1β, S100 proteins may promote Th17, γδ T cell development, or induce IL-17 expression in autoreactive CD8+ T cells, sustaining a potentially important amplification loop mediated by activated neutrophils (197, 338–340).
In sJIA patients, plasma or serum levels of IL-17 were found to be either normal or increased compared to those of HCs (168, 340, 341). Intracellular flow cytometric staining showed that IL-17 was increasingly expressed in circulating γδ T cells of patients with sJIA. Furthermore, an increased number of IL-17-producing T cells in patients was reported (340, 342). Using the sJIA-like mouse model, we showed that IL-17 is a major cytokine driving the disease pathogenesis (343). In analogy, in patients with sJIA, the development of anaemia was linked to IL-17 since a positive correlation was seen between circulating IL-17 and the erythropoiesis signature (344). In addition to the development of anaemia, IL-17 expression may explain multiple disease symptoms including arthritis, fever (e.g. by the release of prostaglandins such as PGE2), and neutrophilia (148, 345–348).
Already since the first reports describing IL-17, an indirect role of the cytokine on neutrophil proliferation and differentiation was demonstrated (281). IL-17 mainly functions on epithelial, endothelial, and stromal cells, inducing the expression of pro-inflammatory cytokines, growth factors, and chemokines that regulate granulopoiesis, recruitment, and life span of new neutrophils (349). IL-6 was the first identified target gene downstream of IL-17, regulating granulopoiesis (350). Furthermore, IL-17 induces the production and release of the two main granulocytic growth factors, namely G-CSF and GM-CSF (281, 351, 352). Since IL-23 is the major regulator of IL-17 release and subsequent G-CSF induction, its pathway is often referred to as the “IL-23-IL-17-G-CSF axis” (353). Remark that this axis also plays a central role during neutrophil homeostasis. Here, phagocytosis of dying neutrophils by specialised macrophages in the tissue i.e. lung, BM, or spleen, may block the secretion of IL-23 and subsequent IL-17-regulated neutrophil release (described above) (112, 354). IL-17 also induces the production of other pro-inflammatory cytokines including TNFα, IL-1β as well as cyclooxygenase 2 (COX2), and inducible nitric oxide synthase (iNOS), which all directly or indirectly regulate the formation of new neutrophils (355). Next, IL-17 also regulates the attraction of neutrophils via the induction of various chemokines i.e. CXCL1 (KC), CXCL2 (MIP2), CXCL6 (GCP-2), or CXCL8 (IL-8) (356). Furthermore, IL-17 can stimulate endothelial expression of P-selectins, E-selectins, and integrin ligands including ICAM-1 and VCAM, enhancing the neutrophil mobilisation (357). Remark that neutrophils themselves can form a positive feedback loop during neutrophil recruitment by releasing pro-inflammatory cytokines and chemokines (8). Unfortunately, no reports exist on the use of IL-17 blocking agents (such as secukinumab, ixekizumab, or bimekizumab) in treating sJIA patients nor on its role in driving neutrophilia.
IL-1β
IL-1β is a cytokine with many pro-inflammatory activities. Several inflammatory cell types including activated monocytes, neutrophils, or macrophages secrete the cytokine via a two-step mechanism. PAMPs or damage-associated molecular patterns (DAMPs) induce the transcription of pro-IL-1β. Subsequent processing into active IL-1β by inflammasomes requires the presence of a second stimulus (358). IL-1β can bind its IL-1R type I (IL-1RI) that, together with the IL-1 receptor accessory protein (IL-1RAcP), induces MyD88-dependent signalling (359).
The pro-inflammatory activity of IL-1β is further regulated by the naturally occurring IL-1 receptor antagonist (IL-1Ra) and by IL-1 receptor type 2 (IL-1R2) (360). IL-1Ra blocks the binding of IL-1 to its signalling receptor. IL-1R2, which is mainly expressed on neutrophils and their precursors, lacks the signalling Toll/IL-1R domain and regulates the pro-inflammatory activity of IL-1β by acting as a decoy receptor (361, 362). The IL-1R2 has a higher affinity for IL-1 than for the IL-1Ra and thus further enhances the anti-inflammatory function of IL-1Ra (363). The surface expression of IL-1R2 is tightly regulated. Anti-inflammatory molecules such as glucocorticoids may augment the surface expression of IL-1R2 and may in part explain the beneficial effects of glucocorticoid treatment in sJIA (364, 365). Pro-inflammatory molecules such as ROS, LPS, TNF-α, leukotriene B4, and fMLF can initiate a rapid proteolytic cleavage of the membrane-bound IL-1R2 by different proteases (364, 366).
Myelopoiesis is stimulated both directly and indirectly upon binding of IL-1β to its receptor. IL-1β itself or in synergy with other growth factors (e.g. G-CSF), can induce the proliferation and differentiation of HSPCs and GMPs. This induction is based on the activation of PU.1 and can be blocked in IL-1R1-deficient mice (367, 368). In naïve conditions, IL-1R1-deficient mice have no defects in myeloid cell numbers (369). Alternatively, IL-1β can indirectly regulate granulopoiesis by modulating the production of neutrophilic growth factors and inflammatory mediators including IL-3, IL-6, G-CSF, and GM-CSF (121, 370–372). More importantly, IL-1β - together with IL-23 - is a potent inducer of IL-17 expression in CD4+ T cells or γδ T cells (337, 373). IL-1β additionally regulates the recruitment of neutrophils by the production of neutrophil attracting chemokines such as CXCL1, CXCL2, or CXCL8 and by the induction of adhesion molecules on endothelial cells (370, 374, 375). Interestingly, IL-1β can also directly prime neutrophils for ROS production or NET formation and may prolong their lifespan (149, 244, 376–378).
Many of the symptoms observed in sJIA patients including fever, rash, thrombocytosis, neutrophilia, and arthritis, can be explained by the increased production of IL-1β (359). However, plasma IL-1β levels are hard to measure and multiple studies failed to show increased plasma or serum IL-1β levels (340, 341, 379–382). Neither was an increased IL-1β gene expression profile observed in patients (383–385). Increased IL-1β production was demonstrated upon stimulation of PBMCs with serum of sJIA patients. However, a reduced IL-1β secretion by monocytes of sJIA patients was reported, suggesting that other cellular sources, such as neutrophils, might play an important role in the production of IL-1β in these patients (168, 218, 386). The successful treatment with IL-1-blocking agents such as anakinra, canakinumab, and rilonacept was the first proof of the importance of IL-1 in sJIA (218, 222, 381, 387, 388). IL-1 blocking therapies are reported to be equally beneficial as first-line therapy, underlining the importance of IL-1β at the disease onset (221, 389). This is in line with the “window of opportunity” that has been proposed by Nigrovic (345). A localised action of IL-1β or activity at low levels might explain the absence of an elevated plasma IL-1β signature (148). Follow-up studies and the identification of single nucleotide polymorphisms (SNPs) in IL-1-related genes have also pointed towards a pathological role for IL-1β in sJIA (158, 168, 218, 384).
In sJIA, the number of neutrophils was rapidly normalised during disease remission and after treatment with anakinra (149). A high (immature) neutrophil count was found to correlate with a good response to anakinra and a short disease duration assuming that the effect of anakinra is mainly due to its effects on neutrophils (149, 168). Indeed, IL-1β has been demonstrated to be an important cytokine in the priming of neutrophils, which could be reverted upon anakinra treatment (149). Also in patients with AOSD, a strongly elevated neutrophil number was associated with an IL-1 gene expression profile and a pronounced upregulation of canakinumab-responsive genes (390).
IL-6
IL-6 can be produced by almost all stromal and immune cells in response to e.g. IL-1β, TNF-α, or TLR ligands. IL-6 binds the IL-6 receptor (IL-6R) which is expressed on a wide variety of cell types and signals in a JAK-dependent way (391, 392).
Although IL-6 is not necessary for maintaining neutrophil homeostasis (393), the cytokine plays a critical role during emergencies (394–396) and may stimulate granulopoiesis in the absence of both G-CSF and GM-CSF (371). Administration of IL-6 induces a biphasic neutrophilia in animals via a rapid mobilization of neutrophils from the marginated pool into the circulation (397–399), followed by an accelerated release of neutrophils from the bone marrow, which is induced by a stimulated myeloid cell differentiation (371, 397, 400, 401). IL-6 deficient mice show an impaired neutrophil response after C. albicans infection (402) and patients treated with the humanised monoclonal anti-IL-6R antibody tocilizumab, show transient neutropenia, all demonstrating the neutrophil mobilising properties of IL-6 (403–408). Interestingly, in C/EBPα KO mice, IL-6 can induce neutrophil differentiation by restoring the G-CSF receptor expression (16, 327).
Classically, IL-6 signals upon binding to its membrane-bound IL-6-receptor alpha subunit (IL-6Rα), resulting in gp130 homodimerization, phosphorylation of STAT3 and STAT1 proteins, and downstream signalling (409). IL-6 can also promote IL-6 trans-signalling in cells that express gp130 but lack the IL-6Rα which requires the binding of IL-6 to the soluble IL-6Rα (sIL-6Rα) into an IL-6/IL-6Rα complex. Neutrophils themselves are an important source of sIL-6Rα. IL-6Rα can proteolytically be cleaved via the TNFα converting enzyme-like enzyme (410) upon stimulation with CRP, CXCL8, C5a, LTB4, or platelet-activating factor (PAF) (410). In this way, neutrophils promote IL-6 trans-signalling in other cell types to favour the resolution of inflammation (411–413). Together with IL-6, the IL-6/IL-R complex activates endothelial cells to secrete monocyte chemoattractant protein-1 (MCP-1) (or CCL2) and induces the expression of adhesion molecules (such as ICAM1 or VCAM) to limit neutrophil accumulation while favouring monocyte recruitment (414, 415). This could explain why in animal models of inflammation, the neutrophilic infiltrate is more dominant in IL-6 KO than in WT animals (412, 416). The recruitment of monocytes is protective in acute models, whereas, during chronic inflammation such as CIA, colitis, or experimental autoimmune encephalomyelitis (EAE), IL-6 predominantly fulfils pro-inflammatory functions by favouring mononuclear-cell accumulation, angioproliferation, B cell maturation, and by promoting the anti-apoptotic properties of T cells (416–420).
Conflicting results regarding the direct role of IL-6 on neutrophil function have been reported (421–426). Recently, it was demonstrated that granulocytes are unable to induce STAT-signalling upon stimulation with IL-6 since the expression of gp130 is lost during the maturation of granulocytes (427). One may therefore speculate that the function attributed to IL-6R in neutrophils rather results from its effects on contaminating cell populations (428).
In sJIA, the gene expression of IL-6 (383, 384) and IL-6 protein levels in serum were strongly elevated in patients compared to HCs (168, 340, 341, 379–381, 429–436). Furthermore, increased IL-6 levels were measured in the synovial fluid of sJIA patients (341). Interestingly, high IL-6 levels were associated with more active joint inflammation (437). Several sJIA symptoms - including fever, thrombocytosis, and growth impairment - can at least partially be explained by the elevated levels of IL-6. IL-6 may be responsible for the microcytic anaemia, by blocking the iron supply to the developing erythroid cells and induces the production of acute-phase proteins (e.g. CRP) in hepatocytes (148, 438–440). The crucial role of IL-6 in sJIA was confirmed by the successful use of the humanised anti-IL-6 receptor antibody tocilizumab (222, 223, 227). Tocilizumab treatment could induce neutropenia and increases the risk of infection in patients. However, these adverse effects were outweighed by the beneficial effect on the sJIA-like features (227). Interestingly, the treatment of sJIA patients with tocilizumab was associated with a significantly different neutrophilic gene expression profile and marked upregulation of genes associated with oxidative phosphorylation (441).
IL-18
IL-18 is a pro-inflammatory cytokine constitutively expressed by monocytes, keratinocytes, and epithelial cells. Like IL-1β, the cytokine is produced in a premature form (pro-IL-18) that requires inflammasome-dependent cleavage by caspase-1 in order to become biologically active (442, 443). IL-18 binds the IL-18 receptor expressed on lymphocytes, DCs, and mesenchymal cells. The cytokine signals like IL-1β in a MyD88-dependent way and exerts its pro-inflammatory action increasing the levels of cell adhesion molecules, including chemokine production, and promoting joint inflammation (443). IL-18 is a potent inducer of IFN-γ and is often referred to as “IFN-γ-inducing factor”. IFN-γ, in turn, regulates the activity of IL-18 via the induction of an IL-18 endogenous inhibitor, the IL-18 binding protein (IL-18BP), thus setting up an auto-inhibition loop (442).
IL-18 has pleiotropic effects on neutrophil activation, including pro-inflammatory cytokine expression, degranulation, and priming of the oxidative burst (1, 444, 445). Since the activation of neutrophils induces inflammasome-mediated IL-18 release, the cytokine can trigger a positive activation loop (446). IL-18 has no direct effect on granulopoiesis, but can stimulate the secretion of CXCL1 and CXCL2, two chemokines involved in the recruitment of neutrophils (447). Indirectly, IL-18 may alter neutrophil homeostasis by inducing IFN-γ in T cells and NK cells (442).
High plasma levels of IL-18 were measured in active sJIA patients and have been proposed as a candidate biomarker (341, 379, 429, 437, 448). In contrast, the levels of IL-18BP were found only moderately increased, insufficient to counteract the high levels of IL-18 (449, 450). Also in patients with inactive disease, moderately increased levels of IL-18 were measured (341, 379, 429, 437, 448). IL-18 is considered as an important cytokine involved in the pathogenesis of sJIA (437). A Phase II clinical trial in which AOSD patients were treated with the IL-18-blocking recombinant IL-18PB (Tadeking alfa) showed a favourable safety profile and demonstrated clinical and laboratory efficacy in 50% of the treated patients. Interestingly, both the number of neutrophils as well as the neutrophil-associated S100A8/A9 and S100A12 protein plasma levels were significantly decreased upon treatment (451).
IFN-γ
IFN-γ is a cytokine that is produced predominantly by activated T cells and NK cells upon stimulation with IL-12 and IL-18 (452–454). The cytokine binds to its specific IFN-γ receptor that signals in a JAK/STAT-dependent way. IFN-γ is a cytokine with both pro- and anti-inflammatory properties.
With respect to myelopoiesis, IFN-γ favours the production of monocytes at the expense of neutrophils (455, 456). Consistently, IFN-γ induces the differentiation of human progenitor cells into monocyts while blocking G-CSF-induced granulopoiesis (457). IFN-γ also inhibits neutrophil recruitment in an indirect way, by blocking the development of Th17 cells and by counteracting IL-17- induced neutrophil-related chemokines (458, 459). These findings explain why IFN-γ-deficient mice are marked by massive granulopoiesis during infection with M. tuberculosis or T. gondii (460, 461). Similarly, the sJIA-like mouse model requires an IFN-γ-deficient background and is hallmarked by a massive neutrophilia (343).
IFN-γ can also directly stimulate neutrophils e.g. by alterering the expression of genes involved in migration, chemotaxis, phagocytosis, or apoptosis (462). IFN-γ-stimulated neutrophils have a prolonged life span, an increased capacity for phagocytosis, oxidative burst, and NET formation, and show an enhanced pro-inflammatory cytokine expression (463, 464). In contrast, IFN-γ inhibits the expression of neutrophil-specific chemokines (i.e. CXCL8/IL-8) and the release of key neutrophil-derived soluble mediators (i.e. MMPs and serine proteases), to counteract inflammation-induced tissue damage (454, 465–467). IFN-γ also induces the expression of PD-L1 on neutrophils, which is involved in the suppression of lymphocyte proliferation (468). Besides, IFN-γ tempts the expression of genes involved in antigen-presentation (209, 462, 463).
In sJIA patients, the role of IFN-γ is incompletely understood. The levels of IFN-γ are moderately increased and are low in comparison to its upstream inducer IL-18 (168, 341, 379). An increased number of IFN-γ producing T cells was found in patients. However, a lower IFN-γ expression was measured in these cells. In vitro stimulation of PBMCs resulted in a decreased IFN-γ production in patients when compared to healthy individuals (340, 342). Likewise, sJIA patients display an absent IFN-γ gene signature in their PBMCs (147, 383, 384), NK cells (379), and synovial tissues (469). NK cells of sJIA patients produced less IFN-γ due to a defective phosphorylation of the IL-18 receptor upon signalling (379, 450). Since monocytes of sJIA patients were able to respond to IFN-γ, a limited in vivo exposure was hypothesized in patients (469). Considering the regulatory effect of IFN-γ on IL-17 activity and downstream G-CSF production, enhanced neutrophilia may be linked to this low IFN-γ exposure. Indeed, using our mouse model, we recently described a G-CSF-driven extramedullary myelopoiesis in the IFN-γ KO mice upon CFA-immunisation (Malengier-Devlies et al., manuscript under revision).
Concluding Remarks
In homeostatic conditions, neutrophil accumulation and activation are tightly regulated via distinct regulatory mechanisms that include neutrophil granulopoiesis, release, storage, extravasation, and clearance. To meet the high demand for new neutrophils during severe (systemic) inflammation, emergency granulopoiesis can be induced by diverse yet partially redundant growth factors and cytokines. Given their antimicrobial properties, capacity to release soluble mediators, and direct cell interactions, this de novo generation of neutrophils might be lifesaving during infectious inflammation. However, in the context of autoimmunity, autoinflammation (including sJIA) or disproportionate infection-induced inflammation (including COVID-19) (470), excessive neutrophil production and activation might be destructive to the host. Results from scRNAseq and CyTOF have greatly improved our understanding of neutrophil ontogenesis in homeostatic conditions. However, additional research is needed to understand neutrophil development in emergency situations and at extramedullary sites. It remains to be established whether extramedullary-derived neutrophils have another functionality or plasticity than neutrophils generated in the BM. It will also be important to understand the functional differences between mature and immature neutrophils that emerge during the emergency granulopoiesis. Next, advanced insights on neutrophil functionality, plasticity, and overlap between diverse pathophysiological situations are warranted. For example, in sJIA, it remains particularly interesting to understand and compare the function and phenotype of neutrophils in the joints and blood. This may help us to understand their role in the pathophysiology and the discrimination between the arthritic and febrile phases of the disease. A better understanding of the different cytokines and growth factors regulating neutrophil homeostasis would enable the development of new targeted therapies that prevent uncontrolled tissue inflammation without impairing the anti-microbial function of neutrophils. Yet we are not able to discriminate between a direct effect of the cytokines and growth factors on the observed disease symptoms or an indirect role via its neutrophil-regulatory properties. Neutrophil depletion studies in the sJIA-like mouse model are an excellent tool to understand the role of neutrophils in the pathophysiology of sJIA. Unfortunately, depletions were incomplete and were followed by a fast rebound of new immature neutrophils (471). In sJIA, we might speculate whether, similar to what has been reported for IL-1β, there would exist a window of opportunity in which G-CSF-, GM-CSF-, IL-17- or IL-18-targeting drugs could be considered (e.g. in patients that do not respond to IL-1 or IL-6 therapies or during the arthritic stage of the disease). Next, we could also envisage studying the clinical effects of drugs targeting neutrophil migration. This could be drugs that block the main chemokine receptors expressed by neutrophils (e.g. CXCR1 and CXCR2), adhesion molecules (e.g. CD11b), or chemoattractants [e.g. IL-8 (CXCL8) in humans or GCP-2 (CXCL6), KC (CXCL1), and MIP-2 (CXCL2) in mice]. In conclusion, sJIA is a unique childhood autoinflammatory immune disorder characterised by massive neutrophilia. Neutrophils in sJIA have a hyperactivated phenotype and are thought to play an important role in the pathophysiology of sJIA. The exact role of neutrophils on sJIA-like features including its regulatory cytokines and growth factors are gradually being revealed but certainly require further investigation.
Author Contributions
BM-D wrote the first draft of the manuscript and designed figures with Biorender software. MM, PP, CW, and PM critically read and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study received funding from the KU Leuven (C1 grant no. C16/17/010) and from FWO-Flanders (GOA3218N and GOC3420N). MM obtained a PhD fellowship supported by the L’Oréal – UNESCO for Women in Science initiative and the FWO-Vlaanderen. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
CW obtained unrestricted grants to KU Leuven from Novartis, Roche, GSK immuno-inflammation and Pfizer.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Our sincere thanks go to Prof. Ghislain Opdenakker who critically reviewed the manuscript.
References
1. Nauseef WM, Borregaard N. Neutrophils at Work. Nat Immunol (2014) 15(7):602–11. doi: 10.1038/ni.2921
2. Opdenakker G, El-asrar AA, Van Damme J. Remnant Epitopes Generating Autoimmunity : From Model to Useful Paradigm. Trends Immunol (2020) 41(5):367–78. doi: 10.1016/j.it.2020.03.004
3. Lima SSS, França MS, Godoi CCG, Martinho GH, de Jesus LA, de Romanelli RMC, et al. Neutropenic Patients and Their Infectious Complications at a University Hospital. Rev Bras Hematol Hemoter (2013) 35(1):18–22. doi: 10.5581/1516-8484.20130009
4. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil Function: From Mechanisms to Disease. Annu Rev Immunol (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
5. Mócsai A. Diverse Novel Functions of Neutrophils in Immunity, Inflammation, and Beyond. J Exp Med (2013) 210(7):1283–99. doi: 10.1084/jem.20122220
6. Kumar V, Sharma A. Neutrophils: Cinderella of Innate Immune System. Int Immunopharmacol (2010) 10(11):1325–34. doi: 10.1016/j.intimp.2010.08.012
7. Borregaard N. Neutrophils , From Marrow to Microbes. Immunity (2010) 33(5):657–70. doi: 10.1016/j.immuni.2010.11.011
8. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-Derived Cytokines: Facts Beyond Expression. Front Immunol (2014) 5:508. doi: 10.3389/fimmu.2014.00508
9. Scapini P, Cassatella MA. Social Networking of Human Neutrophils Within the Immune System. Blood (2017) 124(5):710–20. doi: 10.1182/blood-2014-03-453217
10. Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil Heterogeneity: Implications for Homeostasis and Pathogenesis. Blood (2016) 127(18):2173–81. doi: 10.1182/blood-2016-01-688887
11. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil Kinetics in Health and Disease. Trends Immunol (2010) 31(8):318–24. doi: 10.1016/j.it.2010.05.006
12. Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernández-Calvo G, et al. Co-Option of Neutrophil Fates by Tissue Environments. Cell (2020) 183(5):1282–97. doi: 10.1016/j.cell.2020.10.003
13. Jaillon S, Ponzetta A, Di MD, Santoni A, Bonecchi R, Mantovani A. Neutrophil Diversity and Plasticity in Tumour Progression and Therapy. Nat Rev Cancer (2020) 20(9):485–503. doi: 10.1038/s41568-020-0281-y
14. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, et al. Neutrophils : New Insights and Open Questions. Sci Immunol (2018) 3(30):eaat4579. doi: 10.1126/sciimmunol.aat4579
15. Wirths S, Bugl S, Kopp HG. Neutrophil Homeostasis and its Regulation by Danger Signaling. Blood (2014) 123(23):3563–6. doi: 10.1182/blood-2013-11-516260
16. Manz MG, Boettcher S. Emergency Granulopoiesis. Nat Rev Immunol (2014) 14(5):302–14. doi: 10.1038/nri3660
17. Friedman AD. Transcriptional Regulation of Granulocyte and Monocyte Development. Oncogene (2002) 1:3377–90. doi: 10.1038/sj.onc.1205324
18. Rosmarin AG, Yang Z, Resendes KK. Transcriptional Regulation in Myelopoiesis : Hematopoietic Fate Choice , Myeloid Differentiation , and Leukemogenesis. Exp Hematol (2005) 33:131–43. doi: 10.1016/j.exphem.2004.08.015
19. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and Perivascular Cells Maintain Haematopoietic Stem Cells. Nature (2012) 481:457–62. doi: 10.1038/nature10783
20. Ding L, Morrison SJ. Haematopoietic Stem Cells and Early Lymphoid Progenitors Occupy Distinct Bone Marrow Niches. Nature (2013) 495:231–5. doi: 10.1038/nature11885
21. Cvejic A. Mechanisms of Fate Decision and Lineage Commitment During Haematopoiesis. Immunol Cell Biol (2016) 94:230–5. doi: 10.1038/icb.2015.96
22. Akashi K, He X, Chen J, Iwasaki H, Niu C, Steenhard B, et al. Transcriptional Accessibility for Genes of Multiple Tissues and Hematopoietic Lineages Is Hierarchically Controlled During Early Hematopoiesis. Blood (2003) 101(2):383–90. doi: 10.1182/blood-2002-06-1780
23. Miyamoto T, Iwasaki H, Reizis B, Ye M, Graf T, Weissman IL, et al. Myeloid or Lymphoid Promiscuity as a Critical Step in Hematopoietic Lineage Commitment. Dev Cell (2002) 3:137–47. doi: 10.1016/S1534-5807(02)00201-0
24. Nandakumar SK, Ulirsch JC, Sankaran VG. Advances in Understanding Erythropoiesis : Evolving Perspectives. Br J Haematol (2016) 173:206–18. doi: 10.1111/bjh.13938
25. Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, et al. Distinct Routes of Lineage Development Reshape the Human Blood Hierarchy Across Ontogeny. Sci (80- ) (2016) 351(6269):139. doi: 10.1126/science.aab2116
26. Paul F, Arkin Y, Porse BT, Paul F, Arkin Y, Giladi A, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors Article Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell (2015) 163(7):1663–77. doi: 10.1016/j.cell.2015.11.013
27. Akashi K, Traver D, Miyamoto T. A Clonogenic Common Myeloid Progenitor That Gives Rise to All Myeloid Lineages. Nature (2000) 404:193–7. doi: 10.1038/35004599
28. Warren L, Bryder D, Weissman IL, Quake SR. Transcription Factor Profiling in Individual Hematopoietic Progenitors by Digital RT-PCR. Proc Natl Acad Sci (2006) 103(47):17807–12. doi: 10.1073/pnas.0608512103
29. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature (2014) 505(7484):559–63. doi: 10.1038/nature12820
30. Ward AC, Loeb DM, Soede-bobok AA, Touw IP, Friedman AD. Regulation of Granulopoiesis by Transcription Factors and Cytokine Signals. Leukemia (2000) 14:973–90. doi: 10.1038/sj.leu.2401808
31. Mora-Jensen H, Jendholm J, Fossum A, Porse B, Borregaard N, Theilgaard-mönch K. Technical Advance : Immunophenotypical Characterization of Human Neutrophil Differentiation. J Leukoc Biol (2011) 90:629–34. doi: 10.1189/jlb.0311123
32. Klausen P, Bjerregaard MD, Borregaard N, Cowland JB. End-Stage Differentiation of Neutrophil Granulocytes In Vivo is Accompanied by Up-Regulation of P27kip1 and Down-Regulation of CDK2, CDK4, and CDK6. J Leukoc Biol (2004) 75(March):569–78. doi: 10.1189/jlb.1003474
33. Cowland JB, Borregaard N. Granulopoiesis and Granules of Human Neutrophils. Immunol Rev (2016) 273:11–28. doi: 10.1111/imr.12440
34. Lawrence SM, Corriden R, Nizet V. The Ontogeny of a Neutrophil : Mechanisms of Granulopoiesis and Homeostasis. Microbiol Mol Biol Rev (2018) 82(1):e00057–17. doi: 10.1128/MMBR.00057-17
35. Skokowa J, Cario G, Uenalan M, Schambach A, Germeshausen M, Battmer K, et al. LEF-1 is Crucial for Neutrophil Granulocytopoiesis and its Expression is Severely Reduced in Congenital Neutropenia. Nat Med (2006) 12(10):1191–8. doi: 10.1038/nm1474
36. Guo H, Ma O, Speck NA, Friedman AD. Runx1 Deletion or Dominant Inhibition Reduces Cebpa Transcription via Conserved Promoter and Distal Enhancer Sites to Favor Monopoiesis Over Granulopoiesis. Blood (2012) 119(19):4408–18. doi: 10.1182/blood-2011-12-397091
37. Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR, et al. C-Myc Is a Critical Target for C/EBPa in Granulopoiesis. Mol Cell Biol (2001) 21(11):3789–806. doi: 10.1128/MCB.21.11.3789-3806.2001
38. Wang Q, Friedman AD. CCAAT / Enhancer-Binding Proteins are Required for Granulopoiesis Independent of Their Induction of the Granulocyte Colony-Stimulating Factor Receptor. Blood (2002) 99(8):2776–85. doi: 10.1182/blood.V99.8.2776
39. Obaya AJ, Mateyak MK, Sedivy JM. Mysterious Liaisons : The Relationship Between C-Myc and the Cell Cycle. Oncogene (1999) 18:2934–41. doi: 10.1038/sj.onc.1202749
40. Shahrin NH, Diakiw S, Dent LA, Brown AL, Andrea RJD. Conditional Knockout Mice Demonstrate Function of Klf5 as a Myeloid Transcription Factor. Blood (2016) 128(1):55–9. doi: 10.1182/blood-2015-12-684514
41. Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, et al. Single-Cell Characterization of Haematopoietic Progenitors and Their Trajectories in Homeostasis and Perturbed Haematopoiesis. Nat Cell Biol (2018) 20:836–46. doi: 10.1038/s41556-018-0121-4
42. Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, et al. Single-Cell Analysis of Mixed-Lineage States Leading to a Binary Cell Fate Choice. Nature (2016) 537(29):698–702. doi: 10.1038/nature19348
43. Kurotaki D, Yamamoto M, Nishiyama A, Uno K, Ban T, Ichino M, et al. IRF8 Inhibits C/EBPa Activity to Restrain Mononuclear Phagocyte Progenitors From Differentiating Into Neutrophils. Nat Commun (2014) 5(4978):1–15. doi: 10.1038/ncomms5978
44. Yanez A, Ng MY, Hassanzadeh-kiabi N, Goodridge HS. Regular Article IRF8 Acts in Lineage-Committed Rather Than Oligopotent Progenitors to Control Neutrophil vs Monocyte Production. Blood (2015) 125(9):1452–9. doi: 10.1182/blood-2014-09-600833
45. Bartels M, Govers AM, Fleskens V, Lourenc AR, Pals CE, Vervoort SJ, et al. Acetylation of C/EBPe Is a Prerequisite for Terminal Neutrophil Differentiation. Blood (2015) 125(11):1782–92. doi: 10.1182/blood-2013-12-543850
46. Liu Q, Dong F. Gfi-1 Inhibits the Expression of Eosinophil Major Basic Protein (MBP) During G-CSF-Induced Neutrophilic Differentiation. Int J Hematol (2013) 95(6):640–7. doi: 10.1007/s12185-012-1078-x
47. Sheshachalam A, Srivastava N, Mitchell T, Lacy P, Eitzen G. Granule Protein Processing and Regulated Secretion in Neutrophils. Front Immunol (2014) 5:448. doi: 10.3389/fimmu.2014.00448
48. Bjerregaard MD, Jurlander J, Klausen P, Borregaard N, Cowland JB. The In Vivo Profile of Transcription Factors During Neutrophil Differentiation in Human Bone Marrow. Blood (2003) 101(11):4322–32. doi: 10.1182/blood-2002-03-0835
49. Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion , Trafficking , and Effector Functions. Immunity (2018) 48:364–79. doi: 10.1016/j.immuni.2018.02.002
50. Yamanaka R, Barlow C, Lekstrom-Himes J, Castilla LH, Liu PP, Eckaus M, et al. Impaired Granulopoiesis , Myelodysplasia , and Early Lethality in CCAAT Enhancer Binding Protein E -Deficient Mice. Proc Natl Acad Sci (1997) 94:13187–92. doi: 10.1073/pnas.94.24.13187
51. Clements WK, Kim AD, Ong KG, Moore JC, Lawson ND, Traver D. A Somitic Wnt16/Notch Pathway Specifies Haematopoietic Stem Cells. Nature (2011) 474(9):220–4. doi: 10.1038/nature10107
52. Pellmé S, Mörgelin M, Tapper H, Mellqvist U-H, Dahlgren C, Karlsson A. Localization of Human Neutrophil Interleukin-8 (CXCL-8) to Organelle(s) Distinct From the Classical Granules and Secretory Vesicles. J Leukoc Biol (2006) 79(3):564–73. doi: 10.1189/jlb.0505248
53. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of Neutrophils. Nat Rev Immunol (2019) 19:255–65. doi: 10.1038/s41577-019-0141-8
54. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10 + and Immature CD10 - Neutrophils Present in G-CSF-Treated Donors Display Opposite Effects on T Cells. Blood (2017) 129(10):1343–56. doi: 10.1182/blood-2016-04-713206
55. Kim M, Yang D, Kim M, Kim S, Kim D. A Late-Lineage Murine Neutrophil Precursor Population Exhibits Dynamic Changes During Demand- Adapted Granulopoiesis. Sci Rep (2017) 7:39804. doi: 10.1038/srep39804
56. Zhu YP, Padgett L, Dinh HQ, Marcovecchio P, Blatchley A, Wu R, et al. Identification of an Early Unipotent Neutrophil Progenitor With Pro-Tumoral Activity in Mouse and Human Bone Marrow. Cell Rep (2018) 24(9):2329–2341.e8. doi: 10.1016/j.celrep.2018.07.097
57. Xie X, Shi Q, Wu P, Zhang X, Kambara H, Su J, et al. Single-Cell Transcriptome Profiling Reveals Neutrophil Heterogeneity in Homeostasis and Infection. Nat Immunol (2020) 21(9):119–1133. doi: 10.1038/s41590-020-0736-z
58. Devi S, Wang Y, Chew WK, Lima R, A-gonzález N, Mattar CNZ, et al. Neutrophil Mobilization via Plerixafor- Mediated CXCR4 Inhibition Arises From Lung Demargination and Blockade of Neutrophil Homing to the Bone Marrow. J Exp Med (2013) 210(11):2321–36. doi: 10.1084/jem.20130056
59. Kawaia T, Malechb HL. WHIM Syndrome: Congenital Immune Deficiency Disease. Curr Opin Hematol (2009) 16(1):20–6. doi: 10.1097/MOH.0b013e32831ac557
60. Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC, Louis S. G-CSF Is an Essential Regulator of Neutrophil Trafficking From the Bone Marrow to the Blood. Immunity (2002) 17:413–23. doi: 10.1016/S1074-7613(02)00424-7
61. Kim HK, De La M, Sierra L, Williams CK, Gulino AV, Tosato G. G-CSF Down-Regulation of CXCR4 Expression Identified as a Mechanism for Mobilization of Myeloid Cells. Blood (2006) 108(3):812–20. doi: 10.1182/blood-2005-10-4162
62. Kohler A, De Filippo K, Hasenberg M, Van Den Brandt C, Nye E, Hosking MP, et al. G-CSF – Mediated Thrombopoietin Release Triggers Neutrophil Motility and Mobilization From Bone Marrow via Induction of Cxcr2 Ligands. Blood (2011) 117(16):4349–57. doi: 10.1182/blood-2010-09-308387
63. Starckx S, Van den Steen PE, Wuyts A, Van Damme J, Opdenakker G. Neutrophil Gelatinase B and Chemokines in Leukocytosis and Stem Cell Mobilization. Leuk Lymphoma (2002) 43(2):233–41. doi: 10.1080/10428190290005982
64. Haese AD, Wuyts A, Dillen C, Dubois B, Billiau A, Heremans H, et al. In Vivo Neutrophil Recruitment by Granulocyte Chemotactic Protein-2 Is Assisted by Gelatinase B / MMP-9 in the Mouse. J Interf Cytokine Res (2000) 20:667–74. doi: 10.1089/107999000414853
65. Van Den Steen PE, Wuyts A, Husson SJ, Proost P, Van Damme J, Opdenakker G. Gelatinase B/MMP-9 and Neutrophil Collagenase/MMP-8 Process the Chemokines Human GCP-2/CXCL6, ENA-78/CXCL5 and Mouse GCP-2/LIX and Modulate Their Physiological Activities. Eur J Biochem (2003) 270(18):3739–49. doi: 10.1046/j.1432-1033.2003.03760.x
66. Pruijt JF, Fibbe WE, Laterveer L, Pieters RA, Lindley IJ, Paemen L, et al. Prevention of Interleukin-8-Induced Mobilization of Hematopoietic Progenitor Cells in Rhesus Monkeys by Inhibitory Antibodies Against the Metalloproteinase Gelatinase B (MMP-9). Proc Natl Acad Sci USA (1999) 96(19):10863–8. doi: 10.1073/pnas.96.19.10863
67. Grassi L, Pourfarzad F, Ullrich S, Stunnenberg HG, Martens JHA, Kuijpers TW, et al. Dynamics of Transcription Regulation in Human Bone Marrow Myeloid Differentiation to Mature Blood Neutrophils. Cell Rep (2018) 24(10):2784–94. doi: 10.1016/j.celrep.2018.08.018
68. Doerschuk CM, Beyers N, Coxson HO, Wiggs B, Hogg JC. Comparison of Neutrophil and Capillary Diameters and Their Relation to Neutrophil Sequestration in the Lung. J Appl Physiol (1985) 74(6):3040–5. doi: 10.1152/jappl.1993.74.6.3040
69. Price TH, Dale DC. Neutrophil Preservation : The Effect of Short-Term Storage on In Vivo Kinetics. J Clin Invest (1977) 59(3):475–80. doi: 10.1172/JCI108662
70. Athens JW, Haab OP, Raab SO, Mauer AM, Ashenbrucker H, Cartwright GE, et al. Leukokinetic Studies. IV. The Total Blood, Circulating and Marginal Granulocyte Pools and the Granulocyte Turnover Rate in Normal Subjets. J Clin Invest (1961) 40(6):989–95. doi: 10.1172/JCI104338
71. Bierman HR, Kelly KH, Cordes FL, Byron RL, Polhemus JA, Rappoport S. The Release of Leukocytes and Platelets From the Pulmonary Circulation by Epinephrine. Blood (1952) 7:683–92. doi: 10.1182/blood.V7.7.683.683
72. Bierman HR, Kelly KH, King FW, Petrakis NL. The Pulmonary Circulation as a Source of Leucocytes and Platelets in Man. Sci (80-) (1951) 114(2959):276–7. doi: 10.1126/science.114.2959.276
73. Yipp BG, Kim JH, Lima R, Zbytnuik LD, Petri B, Swanlund N, et al. The Lung is a Host Defense Niche for Immediate Neutrophil-Mediated Vascular Protection. Sci Immunol (2017) 2(10):eaam8929. doi: 10.1126/sciimmunol.aam8929
74. Sadik CD, Luster AD. Lipid-Cytokine-Chemokine Cascades Orchestrate Leukocyte Recruitment in Inflammation. J Leukoc Biol (2012) 91:207–15. doi: 10.1189/jlb.0811402
75. Petri B, Sanz M. Neutrophil Chemotaxis. Cell Tissue Res (2018) 371:425–36. doi: 10.1007/s00441-017-2776-8
76. Metzemaekers M, Gouwy M, Proost P. Neutrophil Chemoattractant Receptors in Health and Disease : Double-Edged Swords. Cell Mol Immunol (2020) 17:433–50. doi: 10.1038/s41423-020-0412-0
77. Schramm R, Thorlacius H. Neutrophil Recruitment in Mast Cell-Dependent Inflammation : Inhibitory Mechanisms of Glucocorticoids. Inflammation Res (2004) 53:644–52. doi: 10.1007/s00011-004-1307-8
78. Kolaczkowska E, Kubes P. Neutrophil Recruitment and Function in Health and Inflammation. Nat Rev Immunol (2013) 13:159–75. doi: 10.1038/nri3399
79. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the Site of Inflammation : The Leukocyte Adhesion Cascade Updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
80. Filippi M. Neutrophil Transendothelial Migration : Updates and New Perspectives. Blood (2019) 133(20):2149–58. doi: 10.1182/blood-2018-12-844605
81. Nourshargh S, Hordijk PL, Sixt M. Breaching Multiple Barriers : Leukocyte Motility Through Venular Walls and the Interstitium. Nat Rev Mol Cell Biol (2010) 11:366–78. doi: 10.1038/nrm2889
82. Morikis VA, Simon SI, Hidalgo A, Alon R, Rossaint J, Simon SI. Neutrophil Mechanosignaling Promotes Integrin Engagement With Endothelial Cells and Motility Within Inflamed Vessels. Front Immunol (2018) 9:1–14. doi: 10.3389/fimmu.2018.02774
83. Maas SL, Soehnlein O, Viola JR. Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front Immunol (2018) 9:2739. doi: 10.3389/fimmu.2018.02739
84. Soehnlein O. Direct and Alternative Antimicrobial Mechanisms of Neutrophil-Derived Granule Proteins. J Membr Biol (2009) 87:1157–64. doi: 10.1007/s00109-009-0508-6
85. Faurschou M, Borregaard N. Neutrophil Granules and Secretory Vesicles in Inflammation. Microbes Infect (2003) 5:1317–27. doi: 10.1016/j.micinf.2003.09.008
86. Cassatella MA. Neutrophil-Derived Proteins : Selling Cytokines by the Pound. Adv Immunol (1999) 73:369–509. doi: 10.1016/S0065-2776(08)60791-9
87. Tecchio C, Cassatella MA. Neutrophil-Derived Chemokines on the Road to Immunity. Semin Immunol (2016) 28(2):119–28. doi: 10.1016/j.smim.2016.04.003
88. Scapini P, Lapinet-vera JA, Cassatella MA. The Neutrophil as a Cellular Source of Chemokines. Immunol Rev (2000) 177:195–203. doi: 10.1034/j.1600-065X.2000.17706.x
89. Sica A, Matsushima K, Van Damme J, Wang M, Poletarutti N, Dejana E, et al. IL-1 Transcriptionally Activates the Neutrophil Chemotactic Factor / IL-8 Gene in Endothelial Cells. Immunology (1990) 199:548–53.
90. Kasama T, Miwa Y, Isozaki T, Odai T, Adachi M, Kunkel SL. Neutrophil-Derived Cytokines: Potential Therapeutic Targets in Inflammation. Curr Drug Targets - Inflammation Allergy (2005) 4(3):273–9. doi: 10.2174/1568010054022114
91. Benigni G, Dimitrova P, Antonangeli F, Sanseviero E, Milanova V, Blom A, et al. CXCR3/CXCL10 Axis Regulates Neutrophil – NK Cell Cross-Talk Determining the Severity of Experimental Osteoarthritis. J Immunol (2017) 198(5):2115–24. doi: 10.4049/jimmunol.1601359
92. Nathan C. Neutrophils and Immunity: Challenges and Opportunities. Nat Rev Immunol (2006) 6:173–82. doi: 10.1038/nri1785
93. Subrahmanyam YVBK, Yamaga S, Prashar Y, Lee HH, Hoe NP, Kluger Y, et al. RNA Expression Patterns Change Dramatically in Human Neutrophils Exposed to Bacteria. Blood (2001) 97(8):2457–68. doi: 10.1182/blood.V97.8.2457
94. Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, Deleo FR. Global Changes in Gene Expression by Human Polymorphonuclear Leukocytes During Receptor-Mediated Phagocytosis : Cell Fate is Regulated at the Level of Gene Expression. Proc Natl Acad Sci (2002) 99(10):6901–6. doi: 10.1073/pnas.092148299
95. Meyer-Hoffert U, Wiedow O. Neutrophil Serine Proteases : Mediators of Innate Immune Responses. Curr Opin Hematol (2011) 18:19–24. doi: 10.1097/MOH.0b013e32834115d1
96. Pekarek BLA, Starr BA, Toledano AY, Schreiber H. Inhibition O F Tumor Growth by Elimination O F Granulocytes. J Exp Med (1995) 181:435–40. doi: 10.1084/jem.181.1.435
97. Tecchio C, Scapini P, Pizzolo G, Cassatella MA. On the Cytokines Produced by Human Neutrophils in Tumors. Semin Cancer Biol (2013) 23(3):159–70. doi: 10.1016/j.semcancer.2013.02.004
98. Puga I, Barra CM, He B, Cassis L, Gentile M, Comerma L, et al. B Cell – Helper Neutrophils Stimulate the Diversification and Production of Immunoglobulin in the Marginal Zone of the Spleen. Nat Immunol (2013) 13(2):170–80. doi: 10.1038/ni.2194
99. Schwaller J, Schneider P, Mhawech-fauceglia P, Mckee T, Myit S, Matthes T, et al. Neutrophil-Derived APRIL Concentrated in Tumor Lesions by Proteoglycans Correlates With Human B-Cell Lymphoma Aggressiveness. Blood (2007) 109(1):331–8. doi: 10.1182/blood-2006-02-001800
100. Huard B, Schneider P, Roosnek E, Huard B, Mckee T, Bosshard C, et al. APRIL Secreted by Neutrophils Binds to Heparan Sulfate Proteoglycans to Create Plasma Cell Niches in Human Mucosa Find the Latest Version : APRIL Secreted by Neutrophils Binds to Heparan Sulfate Proteoglycans to Create Plasma Cell Niches in Human Mucosa. J Clin Invest (2010) 8:2887–95. doi: 10.1172/JCI33760C1
101. Scapini P, Bazzoni F, Cassatella MA. Regulation of B-Cell-Activating Factor (BAFF)/ B Lymphocyte Stimulator (BLyS) Expression in Human Neutrophils. Immunol Lett (2008) 116(1):1–6. doi: 10.1016/j.imlet.2007.11.009
102. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
103. Miralda I, Uriarte SM, Mcleish KR. Multiple Phenotypic Changes Define Neutrophil Priming. Front Cell Infect Microbiol (2017) 7:217. doi: 10.3389/fcimb.2017.00217
104. Adrover M, Fresno C, Crainiciuc G, Adrover M, Fresno C, Crainiciuc G, et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity (2019) 50:390–402. doi: 10.1016/j.immuni.2019.01.002
105. Adrover JM, Nicolás-ávila JA, Hidalgo A. Aging : A Temporal Dimension for Neutrophils. Trends Immunol (2016) 37(5):334–45. doi: 10.1016/j.it.2016.03.005
106. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-Gonzalez N, et al. Rhythmic Modulation of the Hematopoietic Niche Through Neutrophil Clearance. Cell (2013) 153:1025–35. doi: 10.1016/j.cell.2013.04.040
107. Aroca-Crevillén A, Adrover JM, Hidalgo A. Circadian Features of Neutrophil Biology. Front Immunol (2020) 11:576. doi: 10.3389/fimmu.2020.00576
108. Ella K, Attila M. Circadian Regulation of Neutrophils : Control by a Cell-Autonomous Clock or Systemic Factors? Eur J Clin Invest (2018) 48(Suppl 2):e12965. doi: 10.1111/eci.12965
109. Haspel JA, Anafi R, Brown MK, Cermakian N, Depner C, Desplats P, et al. Perfect Timing : Circadian Rhythms , Sleep , and Immunity — an NIH Workshop. JCI Insight (2020) 5(1):e131487. doi: 10.1172/jci.insight.131487
110. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil Ageing is Regulated by the Microbiome. Nature (2015) 525(7570):528–32. doi: 10.1038/nature15367
111. A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic Cells Promote Their Own Clearance and Immune Tolerance Through Activation of the Nuclear Receptor LXR. Immunity (2009) 31:245–58. doi: 10.1016/j.immuni.2009.06.018
112. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of Apoptotic Neutrophils Regulates Granulopoiesis via IL-23 and IL-17. Immunity (2005) 22:285–94. doi: 10.1016/j.immuni.2005.01.011
113. Croker BA, Silke J, Gerlic M. Fight or Flight - Regulation of Emergency Hematopoiesis by Pyroptosis and Necroptosis. Curr Opin Hematol (2016) 22(4):293–301. doi: 10.1097/MOH.0000000000000148
114. Boettcher S, Manz MG. Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol (2017) 38(5):345–57. doi: 10.1016/j.it.2017.01.004
115. Honda T, Uehara T, Matsumoto G, Arai S, Sugano M. Neutrophil Left Shift and White Blood Cell Count as Markers of Bacterial Infection. Clin Chim Acta (2016) 457:46–53. doi: 10.1016/j.cca.2016.03.017
116. Cavallazzi R, Bennin C, Hirani A, Gilbert C, Marik PE. Is the Band Count Useful in the Diagnosis of Infection ? Accuracy Study Critically Ill Patients J Intensive Care Med (2010) 25(6):353–7. doi: 10.1177/0885066610377980
117. Mare TA, Treacher DF, Shankar-Hari M, Beale R, Lewis SM, Chambers DJ, et al. The Diagnostic and Prognostic Significance of Monitoring Blood Levels of Immature Neutrophils in Patients With Systemic Inflammation. Crit Care (2015) 19(1):57. doi: 10.1186/s13054-015-0778-z
118. Pillay J, Kamp VM, Van Hoffen E, Visser T, Tak T, Lammers J, et al. A Subset of Neutrophils in Human Systemic Inflammation Inhibits T Cell Responses Through Mac-1. J Clin Invest (2012) 122: (1):327–36. doi: 10.1172/JCI57990
119. Leliefeld PHC, Pillay J, Vrisekoop N, Heeres M, Tak T, Kox M, et al. Differential Antibacterial Control by Neutrophil Subsets. Blood Adv (2018) 2(11):1344–55. doi: 10.1182/bloodadvances.2017015578
120. Tak T, Wijten P, Heeres M, Pickkers P, Scholten A, Heck AJR, et al. Human CD62L Dim Neutrophils Identified as a Separate Subset by Proteome Pro Fi Ling and In Vivo Pulse-Chase Labeling. Blood (2017) 129(26):3476–85. doi: 10.1182/blood-2016-07-727669
121. Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, et al. C/EBPbeta is Required for “Emergency” Granulopoiesis. Nat Immunol (2006) 7(7):732–9. doi: 10.1038/ni1354
122. Satake S, Hirai H, Hayashi Y, Tamura A, Yao H, Yoshioka S, et al. C/EBP β Is Involved in the Amplification of Early Granulocyte Precursors During Candidemia-Induced “‘Emergency’” Granulopoiesis. J Immunol (2012) 189:4546–55. doi: 10.4049/jimmunol.1103007
123. Hayashi Y, Hirai H, Kamio N, Yao H, Yoshioka S, Miura Y, et al. C/EBPbeta Promotes BCR-ABL-Mediated Myeloid Expansion and Leukemic Stem Cell Exhaustion. Leukemia (2013) 27:619–28. doi: 10.1038/leu.2012.258
124. Hirai H, Yokota A, Tamura A, Sato A, Maekawa T. Non-Steady-State Hematopoiesis Regulated by the C/Ebpβ Transcription Factor. Cancer Sci (2015) 106(7):797–802. doi: 10.1111/cas.12690
125. Bugl S, Wirths S, Radsak MP, Stein P, Andre MC, Mu MR, et al. Steady-State Neutrophil Homeostasis is Dependent on TLR4 / TRIF Signaling. Blood (2013) 121: (5):723–34. doi: 10.1182/blood-2012-05-429589
126. Hirai H, Kamio N, Huang G, Matsusue A, Ogino S, Kimura N. Cyclic AMP Responsive Element Binding Proteins Are Involved in 'Emergency' Granulopoiesis Through the Upregulation of CCAAT / Enhancer Binding Protein Beta. PloS One (2013) 8(1):e54862. doi: 10.1371/journal.pone.0054862
127. Kim M, Granick JL, Kwok C, Walker NJ, Borjesson DL, Curry FE, et al. Neutrophil Survival and C-Kit ε -Progenitor Proliferation in Staphylococcus Aureus – Infected Skin Wounds Promote Resolution. Blood (2011) 117(12):3343–52. doi: 10.1182/blood-2010-07-296970
128. Jones LC, Lin M, Chen S, Krug U, Hofmann W, Lee S, et al. Expression of C / EBPbeta From the C/ebpapha Gene Locus is Sufficient for Normal Hematopoiesis In Vivo. Blood (2002) 99(6):2032–6. doi: 10.1182/blood.V99.6.2032
129. Porse BT, Pedersen TA, Xu X, Lindberg B, Wewer UM. E2F Repression by C/EBPalpha Is Required for Adipogenesis and Granulopoiesis In Vivo. Cell (2001) 107:247–58. doi: 10.1016/S0092-8674(01)00516-5
130. Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, et al. C / EBPalpha Arrests Cell Proliferation Through Direct Inhibition of Cdk2 and Cdk4. Mol Cell (2001) 8:817–28. doi: 10.1016/S1097-2765(01)00366-5
131. Sebastian T, Johnson PF. Stop and Go: Anti-Proliferative and Mitogenic Functions of the Transcription Factor C/EBPbeta. Cell Cycle (2006) 5(9):953–7. doi: 10.4161/cc.5.9.2733
132. Akagi T, Saitoh T, Kelly JO, Akira S, Gombart AF, Koeffler HP. Impaired Response to GM-CSF and G-CSF , and Enhanced Apoptosis in C/EBPbeta-Deficient Hematopoietic Cells. Blood (2008) 111(6):2999–3004. doi: 10.1182/blood-2007-04-087213
133. Zhang H, Nguyen-jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 Controls Myeloid Progenitor Growth During Emergency Granulopoiesis. Blood (2010) 116(14):2462–71. doi: 10.1182/blood-2009-12-259630
134. Hall CJ, Flores MV, Oehlers SH, Sanderson LE, Lam EY, Crosier KE, et al. Infection-Responsive Expansion of the Hematopoietic Stem and Progenitor Cell Compartment in Zebrafish Is Dependent Upon Inducible Nitric Oxide. Cell Stem Cell (2012) 10(2):198–209. doi: 10.1016/j.stem.2012.01.007
135. Kim CH. Homeostatic and Pathogenic Extramedullary Hematopoiesis. J Blood Med (2010) 1:13–9. doi: 10.2147/JBM.S7224
136. Hosseinzadeh M, Omidifar N, Kumar PV. Pulmonary Extramedullary Hematopoiesis in a Patient With Chronic Asthma Resembling Lung Cancer: A Case Report. Case Rep Med (2012) 3–6. doi: 10.1155/2012/231787
137. Koch CA, Li C-Y, Mesa RA, Tefferi A. Nonhepatosplenic Extramedullary Hematopoiesis: Associated Diseases, Pathology, Clinical Course, and Treatment. Mayo Clin Proc (2003) 78(10):1223–33. doi: 10.4065/78.10.1223
138. Johns JL, Christopher MM. Extramedullary Hematopoiesis: A New Look at the Underlying Stem Cell Niche , Theories of Development , and Occurrence in Animals. Vet Pathol (2012) 49(3):508–23. doi: 10.1177/0300985811432344
139. Lefrançais E, Ortiz-muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The Lung is a Site of Platelet Biogenesis and a Reservoir for Haematopoietic Progenitors. Nature (2017) 544:105–9. doi: 10.1038/nature21706
140. Inra CN, Zhou BO, Acar M, Murphy MM, Richardson J, Zhao Z, et al. A Perisinusoidal Niche for Extramedullary Haematopoiesis in the Spleen. Nature (2015) 527(7579):466–71. doi: 10.1038/nature15530
141. Yvan-Charvet L, Ng LG. Granulopoiesis and Neutrophil Homeostasis : A Metabolic , Daily Balancing Act. Trends Immunol (2019) 40(7):598–612. doi: 10.1016/j.it.2019.05.004
142. Robbins CS, Swirski FK. The Multiple Roles of Monocyte Subsets in Steady State and Inflammation. Cell Mol Life Sci (2010) 67:2685–93. doi: 10.1007/s00018-010-0375-x
143. Wu C, Chen M, Zheng L, Wu C, Ning H, Liu M, et al. Spleen Mediates a Distinct Hematopoietic Progenitor Response Supporting Tumor- Promoting Myelopoiesis Find the Latest Version : Spleen Mediates a Distinct Hematopoietic Progenitor Response Supporting Tumor-Promoting Myelopoiesis. J Clin Invest (2018) 128(8):3425–38. doi: 10.1172/JCI97973
144. Alshetaiwi H, Pervolarakis N, Mcintyre LL, Ma D, Rath JA, Nee K, et al. Defining the Emergence of Myeloid-Derived Suppressor Cells in Breast Cancer Using Single-Cell Transcriptomics. Sci Immunol (2020) 5(44):eaay6017. doi: 10.1126/sciimmunol.aay6017
145. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-Induced Tolerance and Immune Suppression Depend on the C/EBPbeta Transcription Factor. Immunity (2010) 32(6):790–802. doi: 10.1016/j.immuni.2010.05.010
146. Lechner MG, Megiel C, Russell SM, Bingham B, Arger N, Woo T, et al. Functional Characterization of Human Cd33 + And Cd11b + Myeloid-Derived Suppressor Cell Subsets Induced From Peripheral Blood Mononuclear Cells Co-Cultured With a Diverse Set of Human Tumor Cell Lines. J Transl Med (2011) 9:90. doi: 10.1186/1479-5876-9-90
147. Fall N, Barnes M, Thornton S, Luyrink L, Olson J, Ilowite NT, et al. Gene Expression Profiling of Peripheral Blood From Patients With Untreated New-Onset Systemic Juvenile Idiopathic Arthritis Reveals Molecular Heterogeneity That May Predict Macrophage Activation Syndrome. Arthritis Rheum (2007) 56(11):3793–804. doi: 10.1002/art.22981
148. Mellins ED, Macaubas C, Grom AA. Pathogenesis of Systemic Juvenile Idiopathic Arthritis : Some Answers , More Questions. Nat Rev Rheumatol (2011) 7(7):416–26. doi: 10.1038/nrrheum.2011.68
149. ter Haar NM, Tak T, Mokry M, Scholman RC, Meerding JM, de Jager W, et al. Reversal of Sepsis-Like Features of Neutrophils by Interleukin-1 Blockade in Patients With Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheumatol (2018) 70(6):943–56. doi: 10.1002/art.40442
150. Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-Onset Still’s Disease. Autoimmun Rev (2014) 13(7):708–22. doi: 10.1016/j.autrev.2014.01.058
151. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, He X, et al. International League of Associations for Rheumatology Classification of Juvenile Idiopathic Arthritis : Second Revision, Edmonton, 2001. J Rheumatol (2004) 31(2):390–2.
152. Woo P. Systemic Juvenile Idiopathic Arthritis: Diagnosis, Management, and Outcome. Nat Clin Pract Rheumatol (2006) 2(1):28–34. doi: 10.1038/ncprheum0084
153. Gurion R, Lehman TJA, Moorthy LN. Systemic Arthritis in Children : A Review of Clinical Presentation and Treatment. J Inflam (2012) 2012:271569. doi: 10.1155/2012/271569
154. Behrens EM, Beukelman T, Gallo L, Spangler J, Rosenkranz M, Arkachaisri T, et al. Evaluation of the Presentation of Systemic Onset Juvenile Rheumatoid Arthritis : Data From the Pennsylvania Systemic Onset Juvenile Arthritis Registry Evaluation of the Presentation of Systemic Onset Juvenile Rheumatoid Arthritis : Data From the Pennsylva. J Rh (2008) 35(2):343–8.
155. Sikora KA, Grom AA. Update on the Pathogenesis and Treatment of Systemic Idiopathic Arthritis. Curr Opin Pediatr (2012) 23(6):640–6. doi: 10.1097/MOP.0b013e32834cba24
156. Jung J, Kim J, Suh C, Kim H. Roles of Interactions Between Toll-Like Receptors and Their Endogenous Ligands in the Pathogenesis of Systemic Juvenile Idiopathic Arthritis and Adult-Onset Still ‘ s Disease. Front Immunol (2020) 11:583513. doi: 10.3389/fimmu.2020.583513
157. Date Y, Seki N, Kamizono S, Higuchi T, Hirata T, Miyata K, et al. Identification of a Genetic Risk Factor for Systemic Juvenile Rheumatoid Arthritis in the 5’-Flanking Region Onf the TNFa Gene and HLA Genes. Arthritis Rheum (1999) 42(12):2577–82. doi: 10.1002/1529-0131(199912)42:12<2577::AID-ANR10>3.0.CO;2-O
158. Stock CJW, Ogilvie EM, Samuel JM, Fife M, Lewis CM, Woo P. Comprehensive Association Study of Genetic Variants in the IL-1 Gene Family in Systemic Juvenile Idiopathic Arthritis. Genes Immun (2008) 9:349–57. doi: 10.1038/gene.2008.24
159. Fife MS, Gutierrez A, Ogilvie EM, Stock CJW, Samuel JM, Thomson W, et al. Novel IL10 Gene Family Associations With Systemic Juvenile Idiopathic Arthritis. Arthritis Res Ther (2006) 8(5):8–12. doi: 10.1186/ar2041
160. Wakil SM, Monies DM, Abouelhoda M, Al-tassan N, Al-dusery H, Naim EA, et al. Association of a Mutation in LACC1 With a Monogenic Form of Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol (2015) 67(1):288–95. doi: 10.1002/art.38877
161. Jørgensen SE, Christiansen M, Høst C, Glerup M, Mahler B, Lausten MM, et al. Systemic Juvenile Idiopathic Arthritis and Recurrent Macrophage Activation Syndrome Due to a CASP1 Variant Causing Inflammasome Hyperactivation. Rheumatology (2020) 59(10):3099–105. doi: 10.1093/rheumatology/keaa242
162. Fishman D, Humphries S, Woo P. The Effect of Novel Polymorphisms in the Interleukin-6 ( IL-6 ) Gene on IL-6 Transcription and Plasma IL-6 Levels , and an Association With Systemic-Onset Juvenile Chronic Arthritis. J Clin Invest (1998) 102(6):1369–76. doi: 10.1172/JCI2629
163. Ogilvie EM, Fife MS, Thompson SD, Twine N, Tsoras M, Moroldo M, et al. The -174g Allele of the Interleukin-6 Gene Confers Susceptibility to Systemic Arthritis in Children. Arthritis Rheum (2003) 48(11):3202–6. doi: 10.1002/art.11300
164. Möller JC, Paul D, Ganser G, Range U, Gahr M, Kelsch R, et al. IL10 Promoter Polymorphisms are Associated With Systemic Onset Juvenile Idiopathic Arthritis (SoJIA). Clin Exp Rheumatol (2010) 28:912–8.
165. Omoyinmi E, Forabosco P, Hamaoui R, Bryant A, Hinks A, Ursu S, et al. Association of the IL-10 Gene Family Locus on Chromosome 1 With Juvenile Idiopathic Arthritis (JIA). PloS One (2012) 7(10):e47673. doi: 10.1371/journal.pone.0047673
166. De Benedetti F, Meazza C, Vivarelli M, Rossi F, Pistorio A, Lamb R, et al. Functional and Prognostic Relevance of the - 173 Polymorphism of the Macrophage Migration Inhibitory Factor Gene in Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheum (2003) 48(5):1398–407. doi: 10.1002/art.10882
167. Lamb R, Thomson W, Ogilvie EM, Donn R. Positive Association of SLC26A2 Gene Polymorphisms With Susceptibility to Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheum (2007) 56(4):1286–91. doi: 10.1002/art.22444
168. Gattorno M, Piccini A, Lasigliè D, Tassi S, Brisca G, Carta S, et al. The Pattern of Response to Anti-Interleukin-1 Treatment Distinguishes Two Subsets of Patients With Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheum (2008) 58(5):1505–15. doi: 10.1002/art.23437
169. Gattorno M, Tassi S, Carta S, Delfino L, Ferlito F, Pelagatti MA, et al. Pattern of Interleukin-1beta Secretion in Response to Lipopolysaccharide and ATP Before and After Interleukin-1 Blockade in Patients With CIAS1 Mutations. Arthritis Rheum (2007) 56(9):3138–48. doi: 10.1002/art.22842
170. Komiya A, Matsui T, Nogi S, Iwata K, Futami H, Takaoka H, et al. Neutrophil CD64 is Upregulated in Patients With Active Adult-Onset Still ‘ s Disease. Scand J Rheumatol (2012) 41(2):156–8. doi: 10.3109/03009742.2011.644325
171. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated Neutrophil Extracellular Traps Limit Inflammation by Degrading Cytokines and Chemokines. Nat Med (2014) 20(5):511–7. doi: 10.1038/nm.3547
172. Wright HL, Moots RJ, Edwards SW. The Multifactorial Role of Neutrophils in Rheumatoid Arthritis. Nat Rev Rheumatol (2014) 10:593–601. doi: 10.1038/nrrheum.2014.80
173. Frangou E, Vassilopoulos D, Boletis J, Boumpas DT. Autoimmunity Reviews An Emerging Role of Neutrophils and NETosis in Chronic Inflammation and Fibrosis in Systemic Lupus Erythematosus (SLE) and ANCA-Associated Vasculitides (AAV): Implications for the Pathogenesis and Treatment. Autoimmun Rev (2019) 18(8):751–60. doi: 10.1016/j.autrev.2019.06.011
174. He Y, Yang FY, Sun EW. Neutrophil Extracellular Traps in Autoimmune Diseases. Chin Midical J (2018) 131(13):1513–9. doi: 10.4103/0366-6999.235122
175. Salemme R, Peralta LN, Meka SH, Pushpanathan N. The Role of NETosis in Systemic Lupus Erythematosus. J Cell Immunol (2019) 1(2):33–42. doi: 10.33696/immunology.1.008
176. De Candia E. NETosis in Arterial and Venous Thrombosis : A One Size Fits All Mechanism? Intern Emerg Med (2017) 12(1):9–11. doi: 10.1007/s11739-016-1600-x
177. Moschonas IC, Tselepis AD. The Pathway of Neutrophil Extracellular Traps Towards Atherosclerosis and Thrombosis. Atherosclerosis (2019) 288:9–16. doi: 10.1016/j.atherosclerosis.2019.06.919
178. Martinod K, Wagner DD. Thrombosis : Tangled Up in NETs. Blood (2014) 123(18):2768–76. doi: 10.1182/blood-2013-10-463646
179. Collison J. Preventing NETosis to Reduce Thrombosis. Nat Rev Rheumatol (2019) 15(6):317. doi: 10.1038/s41584-019-0234-6
180. Berthelot J, Le B, Neel A, Maugars Y. NETosis : At the Crossroads of Rheumatoid Arthritis , Lupus , and Vasculitis. Jt Bone Spine (2017) 84(3):255–62. doi: 10.1016/j.jbspin.2016.05.013
181. Hu X, Xie Q, Mo X, Jin Y. The Role of Extracellular Histones in Systemic-Onset Juvenile Idiopathic Arthritis. Ital Jouranl Pediatr (2019) 45(14):1–9. doi: 10.1186/s13052-019-0605-2
182. Bobek D, Grčević D, Kovačić N, Lukić IK, Jelušić M. The Presence of High Mobility Group Box-1 and Soluble Receptor for Advanced Glycation End-Products in Juvenile Idiopathic Arthritis and Juvenile Systemic Lupus Erythematosus. Pediatr Rheumatol (2014) 12:50. doi: 10.1186/1546-0096-12-50
183. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi: 10.1126/scitranslmed.3001201
184. Brown RA, Henderlight M, Do T, Yasin S, Grom AA, DeLay M, et al. Neutrophils From Children With Systemic Juvenile Idiopathic Arthritis Exhibit Persistent Proinflammatory Activation Despite Long-Standing Clinically Inactive Disease. Front Immunol (2018) 9:2995/full. doi: 10.3389/fimmu.2018.02995/full
185. Ramanathan K, Glaser A, Lythgoe H, Ong J, Beresford MW, Midgley A, et al. Neutrophil Activation Signature in Juvenile Idiopathic Arthritis Indicates the Presence of Low-Density Granulocytes. Rheumatology (2018) 57(3):488–98. doi: 10.1093/rheumatology/kex441/4732527
186. Verweyen EL, Pickering A, Grom AA, Schulert GS. Distinct Gene Expression Signatures Characterize Strong Clinical Responders Versus Nonresponders to Canakinumab in Children With Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol (2021) 73(7):1334–40. doi: 10.1002/art.41640
187. Kessel C, Holzinger D, Foell D. Phagocyte-Derived S100 Proteins in Autoinflammation : Putative Role in Pathogenesis and Usefulness as Biomarkers. Clin Immunol (2013) 147(3):229–41. doi: 10.1016/j.clim.2012.11.008
188. Foell D, Wittkowski H, Kessel C, Lüken A, Weinhage T, Varga G, et al. Proinflammatory S100A12 can Activate Human Monocytes via Toll-Like Receptor 4. Am J Respir Crit Care Med (2013) 187(12):1324–34. doi: 10.1164/rccm.201209-1602OC
189. Foell D, Wittkowski H, Hammerschmidt I, Wulffraat N, Schmeling H, Frosch M, et al. Monitoring Neutrophil Activation in Juvenile Rheumatoid Arthritis By S100A12 Serum Concentrations. Arthritis Rheum (2004) 50(4):1286–95. doi: 10.1002/art.20125
190. Frosch M, Foell D, Ganser G, Roth J. Arthrosonography of Hip and Knee Joints in the Follow Up of Juvenile Rheumatoid Arthritis. Ann Rheum Dis (2003) 62(3):242–4. doi: 10.1136/ard.62.3.242
191. Frosch M, Ahlmann M, Vogl T, Wittkowski H, Wulffraat N, Foell D, et al. The Myeloid-Related Proteins 8 and 14 Complex , a Novel Ligand of Toll-Like Receptor 4, and Interleukin-1beta Form a Positive Feedback Mechanism in Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheum (2009) 60(3):883–91. doi: 10.1002/art.24349
192. Wittkowski H, Frosch M, Wulffraat N, Frühwald R--, Dassmann S, Pham T-H, et al. S100A12 is a Novel Molecular Marker Differentiating Systemic Onset Juvenile Idiopathic Arthritis From Other Causes of Fever of Unknown Origin. Arthritis Rheum (2008) 58(12):3924–31. doi: 10.1002/art.24137
193. Foell D, Wittkowski H, Roth J. Mechanisms of Disease : A ‘ DAMP ‘ View of Inflammatory Arthritis. Nat Clin Pract Rheumatol (2007) 3(7):382–90. doi: 10.1038/ncprheum0531
194. Holzinger D, Foell D, Kessel C. The Role of S100 Proteins in the Pathogenesis and Monitoring of Autoinflammatory Diseases. Mol Cell Pediatr (2018) 5(1):7. doi: 10.1186/s40348-018-0085-2
195. La C, Lê PQ, Ferster A, Goffin L, Spruyt D, Lauwerys B, et al. Serum Calprotectin (S100A8 / A9): A Promising Biomarker in Diagnosis and Up in Different Subgroups of Juvenile Idiopathic Arthritis. RMD Open (2021) 7:e001646. doi: 10.21203/rs.3.rs-138436/v2
196. Kessel C, Fuehner S, Zell J, Zimmermann B, Drewianka S, Brockmeyer S, et al. Calcium and Zinc Tune Autoinflammatory Toll-Like Receptor 4 Signaling by S100A12. J Allergy Clin Immunol (2018) 142(4):1370–3. doi: 10.1016/j.jaci.2018.06.027
197. Kessel C, Hedrich CM, Foell D. Innately Adaptive or Truly Autoimmune : Is There Something Unique About Systemic Juvenile Idiopathic Arthritis ? Arthritis Rheumatol (2020) 72(2):210–9. doi: 10.1002/art.41107
198. Elemam NM, Hannawi S, Maghazachi AA. Role of Chemokines and Chemokine Receptors in Rheumatoid Arthritis. ImmunoTargets Ther (2020) 9:43–56. doi: 10.2147/ITT.S243636
199. Pillinger MH, Abramson SB. The Neutrophil in Rheumatoid Arthritis. Rheum Dis Clin North Am (1995) 21(3):691–714. doi: 10.1016/S0889-857X(21)00463-4
200. Arend WP. The Innate Immune System in Rheumatoid Arthritis. Arthritis Rheum (2001) 44(10):2224–34. doi: 10.1002/1529-0131(200110)44:10<2224::AID-ART384>3.0.CO;2-H
201. Cecchi I, Arias I, Rosa D, Menegatti E, Roccatello D. Neutrophils : Novel Key Players in Rheumatoid Arthritis. Curr Future Ther Targets Autoimmun Rev (2018) 17(11):1138–49. doi: 10.1016/j.autrev.2018.06.006
202. Chakravarti A, Raquil M, Tessier P, Poubelle PE. Surface RANKL of Toll-Like Receptor 4 – Stimulated Human Neutrophils Activates Osteoclastic Bone Resorption. Blood (2009) 114(8):1633–44. doi: 10.1182/blood-2008-09-178301
203. Jarvis JN, Jiang K, Petty HR, Centola M. Neutrophils : The Forgotten Cell in JIA Disease Pathogenesis. Pediatr Rheumatol (2007) 5:13. doi: 10.1186/1546-0096-5-13
204. Hu Z, Jiang K, Barton M, Chen Y, Jarvis JN. Modeling Transcriptional Rewiring in Neutrophils Through the Course of Treated Juvenile Idiopathic Arthritis. Sci Rep (2018) 8:7805. doi: 10.1038/s41598-018-26163-4
205. Metzemaekers M, Malengier-Devlies B, Yu K, Vandendriessche S, Yserbyt J, Matthys P, et al. Synovial Fluid Neutrophils From Patients With Juvenile Idiopathic Arthritis Display a Hyperactivated Phenotype. Arthritis Rheumatol (2021) 73(5):875–84. doi: 10.1002/art.41605
206. Jung TM, Dailey MO. Rapid Modulation of Homing Receptors (Gp90mel-14) Induced by Activators of Protein Kinase C . Receptor Shedding Due to Accelerated Proteolytic Cleavage at the Cell Information About Subscribing to The Journal of Immunology is Online at : Receptor Sheddi. J Immunol (1990) 144:3130–6.
207. Demaret J, Venet F, Friggeri A, Plassais J, Jallades L, Malcus C, et al. Marked Alterations of Neutrophil Functions During Sepsis-Induced Immunosuppression. J Leukoc Biol (2015) 98:1081–90. doi: 10.1189/jlb.4A0415-168RR
208. Kahn J, Ingraham RH, Shirley F, Migaki GI, Kishimoto TK. Membrane Proximal Cleavage of L-Selectin: Identification of the Cleavage Site and a 6-kD Transmembrane Peptide Fragment of L-Selectin. J Cell Biol (1994) 125(2):461–70. doi: 10.1083/jcb.125.2.461
209. Matsushima H, Geng S, Lu R, Okamoto T, Yao Y, Mayuzumi N, et al. Neutrophil Differentiation Into a Unique Hybrid Population Exhibiting Dual Phenotype and Functionality of Neutrophils and Dendritic Cells. Blood (2013) 121(10):1677–89. doi: 10.1182/blood-2012-07-445189
210. Geng S, Matsushima H, Okamoto T, Yao Y, Lu R, Page K, et al. Emergence , Origin , and Function of Neutrophil-Dendritic Cell Hybrids in Experimentally Induced Inflammatory Lesions in Mice. Blood (2013) 121(10):1690–700. doi: 10.1182/blood-2012-07-445197
211. Liu Y, Xia C, Chen J, Fan C, He J. Elevated Circulating Pro-Inflammatory Low-Density Granulocytes in Adult-Onset Still ‘ s Disease. Rheumatology (2021) 60(1):297–303. doi: 10.1093/rheumatology/keaa324
212. Cain DW, Snowden PB, Sempowski GD, Kelsoe G. Inflammation Triggers Emergency Granulopoiesis Through a Density-Dependent Feedback Mechanism. PloS One (2011) 6(5):e19957. doi: 10.1371/journal.pone.0019957
213. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J Leukoc Biol (2009) 86:1065–73. doi: 10.1189/jlb.0609385
214. Gabrilovich DI, Ostrand-rosenberg S, Bronte V. Coordinated Regulation of Myeloid Cells by Tumours. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
215. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of Tumor-Associated Macrophages and Neutrophils. Proc Natl Acad Sci (2012) 109(7):2491–6. doi: 10.1073/pnas.1113744109
216. Fujii W, Ashihara E, Hirai H, Seno T, Yamamoto A, Ishino H. Myeloid-Derived Suppressor Cells Play Crucial Roles in the Regulation of Mouse Collagen-Induced Arthritis. J Immunol (2013) 191:1073–81. doi: 10.4049/jimmunol.1203535
217. Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A Multicentre, Randomised, Double-Blind, Placebo-Controlled Trial With the Interleukin-1 Receptor Antagonist Anakinra in Patients With Systemic-Onset Juvenile Idiopathic Arthritis (ANAJIS Trial). Ann Rheum Dis (2011) 70:747–54. doi: 10.1136/ard.2010.134254
218. Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of Interleukin-1 (IL-1) in the Pathogenesis of Systemic Onset Juvenile Idiopathic Arthritis and Clinical Response to IL-1 Blockade. J Exp Med (2005) 201(9):1479–86. doi: 10.1084/jem.20050473
219. Yokota S, Itoh Y, Morio T, Origasa H, Sumitomo N, Tomobe M, et al. Tocilizumab in Systemic Juvenile Idiopathic Arthritis in a Real-World Clinical Setting : Results From 1 Year of Postmarketing Surveillance Follow-Up of 417 Patients in Japan. Ann Rheum Dis (2016) 75:1654–60. doi: 10.1136/annrheumdis-2015-207818
220. Woo P, Wilkinson N, Prieur A, Southwood T, Leone V, Livermore P, et al. Research Article Open Label Phase II Trial of Single , Ascending Doses of MRA in Caucasian Children With Severe Systemic Juvenile Idiopathic Arthritis : Proof of Principle of the Efficacy of IL-6 Receptor Blockade in This Type of Arthritis and Demonstrati. Arthritis Res (2005) 7(6):1281–8. doi: 10.1186/ar1826
221. ter Haar NM, Van Dijkhuizen EHP, Swart JF, Van Royen-Kerkhof A, Idrissi A, Leek AP, et al. Treatment to Target Using Recombinant Interleukin-1 Receptor Antagonist as First-Line Monotherapy in Onset Systemic Juvenile Idiopathic Arthritis : Results From a Five-Year Follow-Up Study. Arthritis Rheumatol (2019) 71(7):1163–73. doi: 10.1002/art.40865
222. Horneff G, Schulz AC, Klotsche J, Hospach A, Minden K, Foeldvari I, et al. Experience With Etanercept , Tocilizumab and Interleukin-1 Inhibitors in Systemic Onset Juvenile Idiopathic Arthritis Patients From the BIKER Registry. Arthritis Res Ther (2017) 19(1):256. doi: 10.1186/s13075-017-1462-2
223. Tarp S, Amarilyo G, Foeldvari I, Christensen R, Woo JMP, Cohen N, et al. Efficacy and Safety of Biological Agents for Systemic Juvenile Idiopathic Arthritis : A Systematic Review and Meta-Analysis of Randomized Trials. Rheumatology (2016) 55:669–79. doi: 10.1093/rheumatology/kev382
224. Woerner A, Uettwiller F, Melki I, Mouy R, Wouters C. Biological Treatment in Systemic Juvenile Idiopathic Arthritis : Achievement of Inactive Disease or Clinical Remission on a First , Second or Third Biological Agent. RMD Open (2015) 1(1):e000036. doi: 10.1136/rmdopen-2014-000036
225. Ilowite NT, Prather K, Lokhnygina Y, Schanberg LE, Elder M, Milojevic D, et al. Randomized , Double-Blind , Placebo-Controlled Trial of the Efficacy and Safety of Rilonacept in the Treatment of Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol (2014) 66(9):2570–9. doi: 10.1002/art.38699
226. Ruperto N, Brunner HI, Quartier P, Wulffraat N, Horneff G, Brik R, et al. Two Randomized Trials of Canakinumab in Systemic Juvenile Idiopathic Arthritis. N Engl J Med (2012) 367(25):2396–406. doi: 10.1056/NEJMoa1205099
227. De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized Trial of Tocilizumab in Systemic Juvenile Idiopathic Arthritis. N Engl J Med (2012) 367(25):2385–95. doi: 10.1056/NEJMoa1112802
228. Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, et al. Efficacy and Safety of Tocilizumab in Patients With Systemic-Onset Juvenile Idiopathic Arthritis : A Randomised , Double-Blind , Placebo-Controlled , Withdrawal Phase III Trial. Lancet (2008) 371:998–1006. doi: 10.1016/S0140-6736(08)60454-7
229. Sivakumar R. G-CSF: A Friend or Foe? Immunome Res (2014) 01(s2):1–8. doi: 10.4172/1745-7580.S2.007
230. Roberts AW. G-CSF: A Key Regulator of Neutrophil Production, But That’s Not All! Growth Factors (2005) 23(1):33–41. doi: 10.1080/08977190500055836
231. Metcalf D. Hematopoietic Cytokines. Blood (2008) 111(2):485–91. doi: 10.1182/blood-2007-03-079681
232. Nicola NA, Metcalf D, Johnson GR, Burgess AW. Separation of Functionally Distinct Human Granulocyte-Macrophage Colony-Stimulating Factors. Blood (1979) 54(3):614–27. doi: 10.1182/blood.V54.3.614.bloodjournal543614
233. Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired Production and Increased Apoptosis of Neutrophils in Granulocyte Colony-Stimulating Factor Receptor – Deficient Mice. Immunity (1996) 5:491–501. doi: 10.1016/S1074-7613(00)80504-X
234. Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice Lacking Granulocyte Colony-Stimulating Factor Have Chronic Neutropenia, Granulocyte and Macrophage Progenitor Cell Deficiency, and Impaired Neutrophil Mobilization. Blood (1994) 84(6):1737–46. doi: 10.1182/blood.V84.6.1737.1737
235. Richards MK, Liu F, Iwasaki H, Akashi K, Link DC. Pivotal Role of Granulocyte Colony-Stimulating Factor in the Development of Progenitors in the Common Myeloid Pathway. Blood (2003) 102(10):3562–8. doi: 10.1182/blood-2003-02-0593
236. Lord BI, Bronchudt MH, Owenst S, Chang J, Howellt A, Souza L, et al. The Kinetics of Human Granulopoiesis Following Treatment With Granulocyte Colony-Stimulating Factor In Vivo. Proc Natl Acad Sci (1989) 86:9499–503. doi: 10.1073/pnas.86.23.9499
237. Basu S, Hodgson G, Katz M, Dunn AR. Evaluation of Role of G-CSF in the Production , Survival , and Release of Neutrophils From Bone Marrow Into Circulation. Blood (2008) 100(3):854–61. doi: 10.1182/blood.V100.3.854
238. Brühl H, Cohen CD, Linder S, Kretzler M, Mack M. Post-Translational and Cell Type-Specific Regulation of CXCR4 Expression by Cytokines. Eur J Immunol (2003) 33:3028–37. doi: 10.1002/eji.200324163
239. Nagase H, Miyamasu M, Yamaguchi M, Imanishi M, Tsuno NH, Matsushima K, et al. Cytokine-Mediated Regulation of CXCR4 Expression in Human Neutrophils. J Leukoc Biol (2002) 71:711–7.
240. Petit I, Szyper-kravitz M, Nagler A, Lahav M, Peled A, Habler L, et al. G-CSF Induces Stem Cell Mobilization by Decreasing Bone Marrow SDF-1 and Up-Regulating Cxcr4. Nat Immunol (2002) 3(7):687–94. doi: 10.1038/ni813
241. Bajrami B, Zhu H, Kwak HJ, Mondal S, Hou Q, Geng G, et al. G-CSF Maintains Controlled Neutrophil Mobilization During Acute Inflammation by Negatively Regulating CXCR2 Signaling. J Exp Med (2016) 213(10):1999–2018. doi: 10.1084/jem.20160393
242. Spiekermann K, Roesler J, Emmendoerffer A, Elsner J, Welte K. Functional Features of Neutrophils Induced by G-CSF and GM-CSF Treatment: Differential Effects and Clinical Implications. Leukemia (1997) 11(4):466–78. doi: 10.1038/sj.leu.2400607
243. Altznauer F, Martinelli S, Yousefi S, Thürig C, Schmid I, Conway EM, et al. Inflammation-Associated Cell Cycle – Independent Block of Apoptosis by Survivin in Terminally Differentiated Neutrophils. J Exp Med (2004) 199(10):1343–54. doi: 10.1084/jem.20032033
244. Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of Granulocyte Survival and Programmed Cell Death by Cytokines and Bacterial Products. Blood (1992) 80(8):2012–20. doi: 10.1182/blood.V80.8.2012.2012
245. Demetri GD, Griffin JD. Granulocyte Colony-Stimulating Factor and its Receptor. Blood (1991) 78(11):2791–808. doi: 10.1182/blood.V78.11.2791.2791
246. Nicholson SE, Starr R, Novak U, Hilton DJ, Layton JE. Tyrosine Residues in the Granulocyte Colony-Stimulating Factor (G-CSF) Receptor Mediate G-CSF-Induced Differentiation of Murine Myeloid Leukemic (M1) Cells. J Biol Chem (1996) 271(43):26947–53. doi: 10.1074/jbc.271.43.26947
247. Novak U, Ward AC, Hertzog PJ, Hamilton JA, Paradiso L. Aberrant Activation of JAK/STAT Pathway Components in Response to G-CSF, Interferon-A/P and Interferon-Y in NFS-60 Cells. Growth Factors (1996) 13(3–4):251–60. doi: 10.3109/08977199609003226
248. Ward BAC, Hermans MHA, Smith L, Van Aesch YM, Schelen AM, Antonissen C, et al. Tyrosine-Dependent and -Independent Mechanisms of STAT3 Activation by the Human Granulocyte Colony-Stimulating Factor (G-CSF) Receptor Are Differentially Utilized Depending on G-CSF Concentration. Blood (1999) 93(1):113–24. doi: 10.1182/blood.V93.1.113.401k33_113_124
249. Ward AC, van Aesch YM, Schelen AM, Touw IP. Defective Internalization and Sustained Activation of Truncated Granulocyte Colony-Stimulating Factor Receptor Found in Severe Congenital Neutropenia/Acute Myeloid Leukemia. Blood (1999) 93(2):447–58. doi: 10.1182/blood.V93.2.447.402k37_447_458
250. O’Shea JJ, Gadina M, Schreiber RD. Cytokine Signaling in 2002 : New Surprises in the Jak / Stat Pathway. Cell (2002) 109:121–31. doi: 10.1016/S0092-8674(02)00701-8
251. Snow JW, Abraham N, Ma MC, Abbey NW, Herndier B, Goldsmith MA. STAT5 Promotes Multilineage Hematolymphoid Development In Vivo Through Effects on Early Hematopoietic Progenitor Cells. Blood (2002) 99(1):95–101. doi: 10.1182/blood.V99.1.95
252. Moriggl R, Sexl V, Kenner L, Duntsch C, Stangl K, Gingras S, et al. Stat5 Tetramer Formation is Associated With Leukemogenesis. Cancer Cell (2005) 7:87–99. doi: 10.1016/j.ccr.2004.12.010
253. Bunting KD, Bradley HL, Hawley TS, Moriggl R, Sorrentino BP, Ihle JN. Reduced Lymphomyeloid Repopulating Activity From Adult Bone Marrow and Fetal Liver of Mice Lacking Expression of STAT5. Blood (2002) 99(2):479–87. doi: 10.1182/blood.V99.2.479
254. Teglund S, Mckay C, Schuetz E, Van Deursen JM, Stravopodis D, Wang D, et al. Stat5a and Stat5b Proteins Have Essential and Nonessential, or Redundant, Roles in Cytokine Responses. Cell (1998) 93:841–50. doi: 10.1016/S0092-8674(00)81444-0
255. Durbin JE, Simon MC, Levy DE. Targeted Disruption of the Mouse Stat1 Gene Results in Compromised Innate Immunity to Viral Disease. Cell (1996) 84:443–50. doi: 10.1016/S0092-8674(00)81289-1
256. Meraz MA, White JM, Sheehan KCF, Bach EA, Rodig SJ, Dighe AS, et al. Targeted Disruption of the Stat1 Gene in Mice Reveals Unexpected Physiologic Specificity in the JAK – STAT Signaling Pathway. Cell (1996) 84:431–42. doi: 10.1016/S0092-8674(00)81288-X
257. Chakraborty A, Tweardy DJ. Stat3 and G-CSF-Induced Myeloid Differentiation. Leuk Lymphoma (1998) 30(5–6):433–42. doi: 10.3109/10428199809057555
258. Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, et al. STAT3 Governs Distinct Pathways in Emergency Granulopoiesis and Mature Neutrophils. Blood (2017) 108(12):3682–91. doi: 10.1182/blood-2006-02-003012
259. Lee C, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, et al. STAT3 Is a Negative Regulator of Granulopoiesis But Is Not Required for G-CSF-Dependent Differentiation. Immunity (2002) 17:63–72. doi: 10.1016/S1074-7613(02)00336-9
260. Welte T, Zhang SSM, Wang T, Zhang Z, Hesslein DGT, Yin Z, et al. STAT3 Deletion During Hematopoiesis Causes Crohn ‘ s Disease-Like Pathogenesis and Lethality : A Critical Role of STAT3 in Innate Immunity. Proc Natl Acad Sci (2003) 100(4):1879–84. doi: 10.1073/pnas.0237137100
261. Kamezaki K, Shimoda K, Numata A, Haro T, Kakumitsu H, Yoshie M, et al. Roles of Stat3 and ERK in G-CSF Signaling. Stem Cells (2005) 23(2):252–63. doi: 10.1634/stemcells.2004-0173a
262. Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, Dirago L, et al. SOCS3 Is a Critical Physiological Negative Regulator of G-CSF Signaling and Emergency Granulopoiesis. Immunity (2004) 20:153–65. doi: 10.1016/S1074-7613(04)00022-6
263. Zhang D, Zhang P, Wang N, Heterington CJ, Darlington GJ, Tenen DG. Absence of Granulocyte Colony-Stimulating Factor Signaling and Neutrophil Development in CCAAT Enhancer Binding Protein Alpha-Deficient Mice. Proc Natl Acad Sci (1997) 94:569–74. doi: 10.1073/pnas.94.2.569
264. Betsuyaku T, Liu F, Senior RM, Haug JS, Brown EJ, Jones SL, et al. A Functional Granulocyte Colony-Stimulating Factor Receptor is Required for Normal Chemoattractant-Induced Neutrophil Activation. J Clin Invest (1999) 103(6):825–32. doi: 10.1172/JCI5191
265. Sinha S, Zhu QS, Romero G, Corey SJ. Deletional Mutation of the External Domain of the Human Granulocyte Colony-Stimulating Factor Receptor in a Patient With Severe Chronic Neutropenia Refractory to Granulocyte Colony-Stimulating Factor. J Pediatr Hematol Oncol (2003) 25(10):791–6. doi: 10.1097/00043426-200310000-00010
266. Druhan LJ, Ai J, Massullo P, Kindwall-keller T, Ranalli MA, Avalos BR. Novel Mechanism of G-CSF Refractoriness in Patients With Severe Congenital Neutropenia. Blood (2005) 105(2):584–91. doi: 10.1182/blood-2004-07-2613
267. Skokowa J, Klimiankou M, Klimenkova O, Lan D, Gupta K, Hussein K, et al. Interactions Among HCLS1 , HAX1 and LEF1 Proteins are Essential for G-CSFtriggered Granulopoiesis. Nat Med (2012) 18(10):1550–9. doi: 10.1038/nm.2958
268. Zeidler C, Germeshausen M, Klein C, Welte K. Clinical Implications of ELA2-, HAX1-, and G-CSF-Receptor (CSF3R) Mutations in Severe Congenital Neutropenia. Br J Haematol (2009) 144(4):459–67. doi: 10.1111/j.1365-2141.2008.07425.x
269. Basu S, Hodgson G, Zhang HH, Katz M, Quilici C, Dunn AR. “Emergency” Granulopoiesis in G-CSF-Deficient Mice in Response to Candida Albicans Infection. Blood (2000) 95(12):3725–33. doi: 10.1182/blood.V95.12.3725
270. Zhan BY, Lieschke GJ, Grail D, Dunn AR, Cheers C. Essential Roles for Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and G-CSF in the Sustained Hematopoietic Response of Listeria Monocytogenes –Infected Mice. Blood (1998) 91(3):863–9. doi: 10.1182/blood.V91.3.863.863_863_869
271. Seymour JF, Lieschke GJ, Grail D, Quilici C, Hodgson G, Dunn AR. Mice Lacking Both Granulocyte Colony-Stimulating Factor (CSF) and Granulocyte-Macrophage CSF Have Impaired Reproductive Capacity, Perturbed Neonatal Granulopoiesis, Lung Disease, Amyloidosis, and Reduced Long-Term Survival. Blood (1997) 90(8):3037–49. doi: 10.1182/blood.V90.8.3037
272. Hibbs ML, Quilici C, Kountouri N, John F, Armes JE, Burgess AW, et al. Mice Lacking Three Myeloid Colony-Stimulating Factors (G-CSF, GM-CSF, and M-CSF) Still Produce Macrophages and Granulocytes and Mount an Inflammatory Response in a Sterile Model of Peritonitis. J Immunol (2007) 178:6435–43. doi: 10.4049/jimmunol.178.10.6435
273. Boettcher S, Ziegler P, Schmid MA, Takizawa H, Van Rooijen N, Kopf M, et al. Cutting Edge: LPS-Induced Emergency Myelopoiesis Depends on TLR4-Expressing Nonhematopoietic Cells. J Immunol (2012) 188:5824–8. doi: 10.4049/jimmunol.1103253
274. Tamura M, Hattori K, Nomura H, Oheda M, Kubota N, Imazeki I, et al. Induction of Neutrophilic Granulocytosis in Mice by Administration of Purified Human Native Granulocyte Colony-Stimulating Factor (G-CSF). Biochem Biophys Res Commun (1987) 142(2):454–60. doi: 10.1016/0006-291X(87)90296-8
275. Dührsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D. Effects of Recombinant Human Granulocyte Colony-Stimulating Factor on Hematopoietic Progenitor Cells in Cancer Patients. Blood (1988) 72(6):2074–81. doi: 10.1182/blood.V72.6.2074.2074
276. Bronchud MH, Scarffe JH, Thatcher N, Crowther D, Souza LM, Alton NK, et al. Phase I / II Study of Recombinant Human Granulocyte Colony-Stimulating Factor in Patients Receiving Intensive Chemotherapy for Small Cell Lung Cancer. Br J Cancer (1987) 56(6):809–13. doi: 10.1038/bjc.1987.295
277. Gabrilove JL, Jakubowski A, Scher H, Sternberg C, Wong G, Grous J, et al. Effect of Granulocyte Colony-Stimulating Factor on Neutropenia and Associated Morbidity Due to Chemotherapy for Transitional-Cell Carcinoma of the Urothelium. N Engl J Med (1988) 318:1414–22. doi: 10.1056/NEJM198806023182202
278. Mehta HM, Malandra M, Corey SJ. G-CSF. And GM-CSF in Neutropenia. J Immunol (2016) 195(4):1341–9. doi: 10.4049/jimmunol.1500861
279. Anasetti C, Logan BR, Lee SJ, Waller EK, Weisdorf DJ, Wingard JR, et al. Peripheral-Blood Stem Cells Versus Bone Marrow From Unrelated Donors. N Engl J Med (2012) 367(16):1487–96. doi: 10.1056/NEJMoa1203517
280. Sano E, Ohashi K, Sato Y, Kashiwagi M, Joguchi A, Naruse N. A Possible Role of Autogenous IFN-B for Cytokine Productions in Human Fibroblasts. J Cell Biochem (2007) 100:1459–76. doi: 10.1002/jcb.21128
281. Fossiez F, Djossou O, Chornarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T Cell Interleukin-17 Induces Stromal Cells to Produce Proinflammatory and Hematopoietic Cytokines. J Exp Med (1996) 183:2593–260. doi: 10.1084/jem.183.6.2593
282. Panopoulos AD, Watowich SS. Granulocyte Colony-Stimulating Factor : Molecular Mechanisms of Action During Steady State and ‘Emergency’ Hematopoiesis. Cytokine (2008) 42:277–88. doi: 10.1016/j.cyto.2008.03.002
283. Watari K, Ozawa VK, Tajika K, Tojo A, Tani K, Nagata S, et al. Production of Human Granulocyte Colony Stimulating Factor by Various Kinds of Stromal Cells In Vitro Detected by Enzyme Immunoassay and In Situ Hybridization. Stem Cells (1994) 12:416–23. doi: 10.1002/stem.5530120409
284. Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, et al. A Key Role for G-CSF – Induced Neutrophil Production and Trafficking During Inflammatory Arthritis. Blood (2008) 112(13):5193–202. doi: 10.1182/blood-2008-02-139535
285. Kawano Y, Fukui C, Shinohara M, Wakahashi K, Ishii S, Suzuki T, et al. G-CSF-Induced Sympathetic Tone Provokes Fever and Primes Antimobilizing Functions of Neutrophils via PGE 2. Blood (2017) 129(5):587–97. doi: 10.1182/blood-2016-07-725754
286. Chow A, Lucas D, Hidalgo A, Méndez-ferrer S, Hashimoto D, Scheiermann C, et al. Bone Marrow CD169+ Macrophages Promote the Retention of Hematopoietic Stem and Progenitor Cells in the Mesenchymal Stem Cell Niche. J Exp Med (2010) 208(2):261–71. doi: 10.1084/jem.20101688
287. Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF Potently Inhibits Osteoblast Activity and CXCL12 mRNA Expression in the Bone Marrow. Blood (2005) 106(9):3020–7. doi: 10.1182/blood-2004-01-0272
288. Schlahsa L, Jaimes Y, Blasczyk R, Figueiredo C. Granulocyte-Colony-Stimulatory Factor: A Strong Inhibitor of Natural Killer Cell Function. Transfusion (2011) 51(2):293–305. doi: 10.1111/j.1537-2995.2010.02820.x
289. Morris KT, Castillo EF, Ray AL, Weston LL, Nofchissey RA, Hanson JA, et al. Anti-G-CSF Treatment Induces Protective Tumor Immunity in Mouse Colon Cancer by Promoting Protective NK Cell, Macrophage and T Cell Responses. Oncotarget (2015) 6(26):22338–47. doi: 10.18632/oncotarget.4169
290. Vandenhaute J, Wouters CH, Matthys P. Natural Killer Cells in Systemic Autoinflammatory Diseases : A Focus on Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome. Front Immunol (2020) 10:3089. doi: 10.3389/fimmu.2019.03089
291. Hamilton JA. GM-CSF in Inflammation and Autoimmunity. Trends Immunol (2002) 23(8):403–8. doi: 10.1016/S1471-4906(02)02260-3
292. Fleetwood AJ, Cook AD, Hamilton JA. Functions of Granulocyte-Macrophage Colony-Stimulating Factor. Crit Rev Immunol (2005) 25(5):405–28. doi: 10.1615/CritRevImmunol.v25.i5.50
293. Fukuzawa H, Sawada M, Kayahara T, Morita-fujisawa Y, Suzuki K, Seno H, et al. Identification of GM-CSF in Paneth Cells Using Single-Cell RT-PCR. Biochem Biophys Res Commun (2003) 312:897–902. doi: 10.1016/j.bbrc.2003.11.009
294. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The Encephalitogenicity of Th17 Cells is Dependent on IL-1- and IL-23-Induced Production of the Cytokine GM-CSF. Nat Immunol (2011) 12(6):568–75. doi: 10.1038/ni.2031
295. Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti T-D. Inflammasome-Derived IL-1 β Regulates the Production of GM-CSF by CD4 + T Cells and γ δ T Cells. J Immunol (2012) 188:3107–15. doi: 10.4049/jimmunol.1103308
296. Duhen T, Campbell DJ. IL-1 β Promotes the Differentiation of Polyfunctional Human CCR6 + CXCR3 + Th1/17 Cells That Are Specific for Pathogenic and Commensal Microbes. J Immunol (2014) 193:120–9. doi: 10.4049/jimmunol.1302734
297. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgt Drives Production of the Cytokine GM-CSF in Helper T Cells , Which is Essential for the Effector Phase of Autoimmune Neuroinflammation. Nat Immunol (2011) 12(6):560–657. doi: 10.1038/ni.2027
298. Ozawa H, Aiba S, Nakagawa S, Tagami H. Interferon-γ and Interleukin-10 Inhibit Antigen Presentation by Langerhans Cells for T Helper Type 1 Cells by Suppressing Their CD80 (B7-1) Expression. Eur J Immunol (1996) 26(3):648–52. doi: 10.1002/eji.1830260321
299. Jansen JH, Wientjens GJ, Fibbe WE, Willemze R, Kluin-Nelemans HC. Inhibition of Human Macrophage Coloy Formation by Interleukin 4. J Exp Med (1989) 170(2):577–82. doi: 10.1084/jem.170.2.577
300. Sagawa K, Mochizuki M, Sugita S, Nagai K, Sudo T, Itoh K. Suppression by IL-10 and IL-4 of Cytokine Production Induced by Two-Way Autologous Mixed Lymphocyte Reaction. Cytokine (1996) 8(6):501–6. doi: 10.1006/cyto.1996.0068
301. Adcock IM, Caramori G. Cross-Talk Between Pro-Inflammatory Transcription Factors and Glucocorticoids. Immunol Cell Biol (2001) 79:376–84. doi: 10.1046/j.1440-1711.2001.01025.x
302. Gasson JC. Molecular Physiology of Granulocyte-Macrophage Colony-Stimulating Factor. Blood (1991) 77(6):1131–45. doi: 10.1182/blood.V77.6.1131.1131
303. Lang RA, Metcalf D, Cuthbertson FA, Lyons I, Stanley E, Kelso A, et al. Transgenic Mice Expressing a Hemopoietic Growth Factor Gene (GM-CSF) Develop Accumulations of Macrophages, Blindness, and a Fatal Syndrome of Tissue Damage. Cell (1987) 51:675–86. doi: 10.1016/0092-8674(87)90136-X
304. Metcalf D, Begley CG, Williamson DJ, Nice EC, De Lamarter J, Mermod JJ, et al. Hemopoietic Responses in Mice Injected With Purified Recombinant Murine GM-CSF. Exp Hematol (1987) 15(1):1–9.
305. Burgess AW, Camakaris J, Metcalf D. Purification and Properties of Colony-Stimulating From Mouse Lung-Conditioned Medium. J Biol Chem (1977) 252(6):1998–2003. doi: 10.1016/S0021-9258(18)71855-3
306. Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-scott J, Parker MW, et al. The Granulocyte-Macrophage Colony-Stimulating Factor Receptor : Linking its Structure to Cell Signaling and its Role in Disease. Blood (2009) 114(7):1289–98. doi: 10.1182/blood-2008-12-164004
307. Hansen G, Hercus TR, Mcclure BJ, Stomski FC, Dottore M, Powell J, et al. The Structure of the GM-CSF Receptor Complex Reveals a Distinct Mode of Cytokine Receptor Activation. Cell (2008) 134:496–507. doi: 10.1016/j.cell.2008.05.053
308. Hamilton JA, Achuthan A. Colony Stimulating Factors and Myeloid Cell Biology in Health and Disease. Trends Immunol (2013) 34(2):81–9. doi: 10.1016/j.it.2012.08.006
309. Uchida K, Beck DC, Yamamoto T, Berclaz P-Y, Abe S, Staudt MK, et al. GM-CSF Autoantibodies and Neutrophil Dysfunction in Pulmonary Alveolar Proteinosis. N Engl J Med (2007) 356(6):567–79. doi: 10.1056/NEJMoa062505
310. Sakagami T, Uchida K, Suzuki T, Carey BC, Wood RE, Wert SE, et al. Human GM-CSF Autoantibodies and Reproduction of Pulmonary Alveolar Proteinosis. N Engl J Med (2014) 361(27):2679–81. doi: 10.1056/NEJMc0904077
311. van Pelt LJ, de Craen AJM, Langeveld NE, Weening RS. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) Ameliorates Chemotherapy- Induced Neutropenia in Children With Solid Tumors. Pediatr Hematol Oncol (1997) 14:539–45. doi: 10.3109/08880019709030910
312. Shiomi A, Usui T. Pivotal Roles of GM-CSF in Autoimmunity and Inflammation. Mediators Inflammation (2015) 2015:568543. doi: 10.1155/2015/568543
313. van Nieuwenhuijze A, Koenders M, Roeleveld D, Sleeman MA, van den Berg W, Wicks IP. GM-CSF as a Therapeutic Target in Inflammatory Diseases. Mol Immunol (2013) 56(4):675–82. doi: 10.1016/j.molimm.2013.05.002
314. Okuda K, Foster R, Griffin JD. Signaling Domains of the Beta C Chain of the GM-CSF/IL-3/IL-5 Receptor. Ann N Y Acad Sci (1999) 872:305–13. doi: 10.1111/j.1749-6632.1999.tb08474.x
315. Blalock WL, Chang F, Hoyle PE, Wang X, Algate PA, Franklin RA, et al. Signal Transduction , Cell Cycle Regulatory , and Anti-Apoptotic Pathways Regulated by IL-3 in Hematopoietic Cells : Possible Sites for Intervention With Anti-Neoplastic Drugs. Leukemia (1999) 13:1109–66. doi: 10.1038/sj.leu.2401493
316. van de Laar L, Coffer PJ, Woltman AM. Regulation of Dendritic Cell Development by GM-CSF : Molecular Control and Implications for Immune Homeostasis and Therapy. Blood (2012) 119(15):3383–93. doi: 10.1182/blood-2011-11-370130
317. Matsumura I, Kitamura T, Wakao H, Tanaka H, Albanese C, Downward J, et al. Transcriptional Regulation of the Cyclin D1 Promoter by STAT5 : Its Involvement in Cytokine-Dependent Growth of Hematopoietic Cells. EMBO J (1999) 18(5):1367–77. doi: 10.1093/emboj/18.5.1367
318. Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF)-Induced STAT5 Activation and Target-Gene Expression During Human Monocyte / Macrophage Differentiation. J Leukoc Biol (2002) 71:511–9.
319. Mui AL, Wakao H, Farrell AO, Harada N, Miyajima A. Lnterleukin-3, Granulocyte-Macrophage Colony Stimulating and Interleukin-5 Transduce Signals Through Two STAT5 Homologs. EMBO J (1995) 14(6):1166–75. doi: 10.1002/j.1460-2075.1995.tb07100.x
320. Choi JK, Kim KH, Park H, Park SR, Choi BH. Granulocyte Macrophage-Colony Stimulating Factor Shows Anti-Apoptotic Activity in Neural Progenitor Cells via JAK / STAT5-Bcl-2 Pathway. Apoptosis (2011) 16:127–34. doi: 10.1007/s10495-010-0552-2
321. Katz S, Zsiros V, Dóczi N, Kiss AL. Inflammation-Induced Epithelial-To-Mesenchymal Transition and GM-CSF Treatment Stimulate Mesenteric Mesothelial Cells to Transdifferentiate Into Macrophages. Inflammation (2018) 41(5):1825–34. doi: 10.1007/s10753-018-0825-4
322. Yang T, Chang P, Kuo T, Lu S. Electronegative L5-LDL Induces the Production of G-CSF and GM-CSF in Human Macrophages Through LOX-1 Involving NFkB and ERK2 Activation. Atherosclerosis (2017) 267:1–9. doi: 10.1016/j.atherosclerosis.2017.10.016
323. Stanley E, Lieschke GJ, Grail D, Metcalff D, Hodgson G, Gall JAM, et al. Granulocyte/macrophage Colony-Stimulating Factor-Deficient Mice Show No Major Perturbation of Hematopoiesis But Develop a Characteristic Pulmonary Pathology. Proc Natl Acad Sci (1994) 91:5592–6. doi: 10.1073/pnas.91.12.5592
324. Kingston D, Schmid MA, Onai N, Obata-onai A, Baumjohann D, Manz MG. The Concerted Action of GM-CSF and Flt3-Ligand on In Vivo Dendritic Cell Homeostasis. Blood (2009) 114(4):835–43. doi: 10.1182/blood-2009-02-206318
325. Zhan Y, Cheers C. Haemopoiesis in Mice Genetically Lacking Granulocyte-Macrophage Colony Stimulating Factor During Chronic Infection With Mycobacterium Avium. Immunol Cell Biol (2000) 78:118–23. doi: 10.1046/j.1440-1711.2000.00891.x
326. Zhang P, Iwama A, Datta MW, Darlington GJ, Link DC, Tenen DG. Upregulation of Interleukin 6 and Granulocyte Colony-Stimulating Factor Receptors by Transcription Factor CCAAT Enhancer Binding Protein Alpha (C/EBP Alpha) is Critical for Granulopoiesis. J Exp Med (1998) 188(6):1173–84. doi: 10.1084/jem.188.6.1173
327. Collins SJ, Ulmer J, Purton LE, Darlington G. Multipotent Hematopoietic Cell Lines Derived From C/EBPa (-/-) Knockout Mice Display Granulocyte Macrophage – Colony-Stimulating Factor, Granulocyte – Colony-Stimulating Factor, and Retinoic Acid – Induced Granulocytic Differentiation. Blood (2001) 96(8):2382–8. doi: 10.1182/blood.V98.8.2382
328. Zhang P, Nelson E, Radomska HS, Iwasaki-Arai J, Akashi K, Friedman AD, et al. Induction of Granulocytic Differentiation by 2 Pathways. Blood (2002) 99(12):4406–12. doi: 10.1182/blood.V99.12.4406
329. Lotfi N, Thome R, Rezaei N, Zhang G, Rezaei A. Roles of GM-CSF in the Pathogenesis of Autoimmune Diseases : An Update. Front Immunol (2019) 10:1265. doi: 10.3389/fimmu.2019.01265
330. Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection From Collagen-Induced Arthritis in Granulocyte-Macrophage Colony-Stimulating Factor-Deficient Mice. J Immunol (1998) 161(7):3639–44.
331. McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte Macrophage Colony-Stimulating Factor: A New Putative Therapeutic Target in Multiple Sclerosis. J Exp Med (2001) 194(7):873–82. doi: 10.1084/jem.194.7.873
332. Bozinovski S, Jones JE, Vlahos R, Hamilton JA, Anderson GP. Granulocyte / Macrophage-Colony-Stimulating Factor (GM-CSF) Regulates Lung Innate Immunity to Lipopolysaccharide Through Akt / Erk Activation of NFkB and AP-1 In Vivo. J Biol Chem (2002) 277(45):42808–14. doi: 10.1074/jbc.M207840200
333. Piper C, Pesenacker AM, Bending D, Thirugnanabalan B, Varsani H, Wedderburn LR, et al. T Cell Expression of Granulocyte-Macrophage Colony-Stimulating Factor in Juvenile Arthritis Is Contingent Upon Th17 Plasticity. Arthritis Rheumatol (2014) 66(7):1955–60. doi: 10.1002/art.38647
334. Gattorno M, Facchetti P, Ghiotto F, Vignola S, Buoncompagni A, Prigione I. Synovial Fluid T Cell Clones From Oligoarticular Juvenile Arthritis Patients Display a Prevalent Th1 / Th0-Type Pattern of Cytokine Secretion Irrespective of Immunophenotype. Clin Exp Immunol (1997) 109:4–11. doi: 10.1046/j.1365-2249.1997.4331330.x
335. Bunte K, Beikler T. Th17 Cells and the IL-23 / IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int J Mol Sci (2019) 20(14):3394. doi: 10.3390/ijms20143394
336. Cua DJ, Tato CM. Innate IL-17-Producing Cells : The Sentinels of the Immune System. Nat Rev Immunol (2010) 10(7):479–89. doi: 10.1038/nri2800
337. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 Induce Innate IL-17 Production From Gamma Delta T Cells , Amplifying Th17 Responses and Autoimmunity. Immunity (2009) 31(2):331–41. doi: 10.1016/j.immuni.2009.08.001
338. Loser K, Vogl T, Voskort M, Lueken A, Kupas V, Nacken W, et al. The Toll-Like Receptor 4 Ligands Mrp8 and Mrp14 are Crucial in the Development of Autoreactive CD8 + T Cells. Nat Med (2010) 16(6):713–8. doi: 10.1038/nm.2150
339. Reinhardt K, Foell D, Vogl T, Wittkowski H, Fend F, Federmann B, et al. Monocyte-Induced Development of Th17 Cells and the Release of S100 Proteins Are Involved in the Pathogenesis of Graft-Versus-Host Disease. J Immunol (2020) 193:3355–65. doi: 10.4049/jimmunol.1400983
340. Kessel C, Lippitz K, Weinhage T, Hinze C, Wittkowski H, Holzinger D, et al. Proinflammatory Cytokine Environments Can Drive Interleukin-17 Overexpression by γ/δ T Cells in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol (2017) 69(7):1480–94. doi: 10.1002/art.40099
341. de Jager W, Hoppenreijs P, Wulffraat NMN, Wedderburn LLR, Kuis W, Prakken BJB, et al. Blood and Synovial Fluid Cytokine Signatures in Patients With Juvenile Idiopathic Arthritis: A Cross-Sectional Study. Ann Rheum Dis (2008) 66(5):589–98. doi: 10.1136/ard.2006.061853
342. Omoyinmi E, Hamaoui R, Pesenacker A, Nistala K, Moncrieffe H, Ursu S, et al. Th1 and Th17 Cell Subpopulations are Enriched in the Peripheral Blood of Patients With Systemic Juvenile Idiopathic Arthritis. Rheumatology (2012) 51(10):1881–6. doi: 10.1093/rheumatology/kes162
343. Avau A, Mitera T, Put K, Brisse E, Filtjens J, Uyttenhove C, et al. Systemic Juvenile Idiopathic Arthritis – Like Syndrome in Mice Following Stimulation of the Immune System With Freund ‘ s Complete Adjuvant. Arthritis Rheumatol (2014) 66(5):1340–51. doi: 10.1002/art.38359
344. Hinze CH, Fall N, Thornton S, Mo JQ, Aronow BJ, Layh-Schmitt G, et al. Immature Cell Populations and an Erythropoiesis Gene-Expression Signature in Systemic Juvenile Idiopathic Arthritis: Implications for Pathogenesis. Arthritis Res Ther (2010) 12(3):R123. doi: 10.1186/ar3061
345. Nigrovic PA. Is There a Window of Opportunity for Treatment of Systemic Juvenile Idiopathic Arthritis ? Arthritis Rheumatol (2014) 66(6):1405–13. doi: 10.1002/art.38615
346. Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The Role of IL-17 and Related Cytokines in Inflammatory Autoimmune Diseases. Mediators Inflammation (2017) 2017:3908061. doi: 10.1155/2017/3908061
347. Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 Family and IL-17 Receptors. Cytokine Growth Factor Rev (2003) 14:155–74. doi: 10.1016/S1359-6101(03)00002-9
348. Roark CL, Simonian PL, Fontenot AP, Born WK, Brien RLO. Gd T Cells : An Important Source of IL-17. Curr Opin Immunol (2008) 20:353–7. doi: 10.1016/j.coi.2008.03.006
349. Kolls JK, Linde A. Interleukin-17 Family Members. Immunity (2004) 21(4):467–76. doi: 10.1016/j.immuni.2004.08.018
350. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirus Saimiri Encodes a New Cytokine, IL-17, Which Binds to a Novel Cytokine Receptor. Immunity (1995) 3(6):811–21. doi: 10.1016/1074-7613(95)90070-5
351. Laan M, Prause O, Miyamoto M, Sjo M, Hyto AM. A Role of GM-CSF in the Accumulation of Neutrophils in the Airways Caused by IL-17 and TNF-A. Eur Respir J (2003) 21:387–93. doi: 10.1183/09031936.03.00303503
352. Schwarzenberger P, Huang W, Ye P, Manuel M, Zhang Z, Bagby G, et al. Requirement of Endogenous Stem Cell Factor and Granulocyte-Colony-Stimulating Factor for IL-17-Mediated Granulopoiesis. J Immunol (2000) 164:4783–9. doi: 10.4049/jimmunol.164.9.4783
353. Iwakura Y, Ishigame H. The IL-23/IL-17 Axis in Inflammation. J Clin Invest (2006) 116(5):1218–22. doi: 10.1172/JCI28508
354. Smith E, Zarbock A, Stark MA, Burcin L, Bruce AC, Foley P, et al. IL-23 Is Required for Neutrophil Homeostasis in Normal and Neutrophilic Mice. J Immunol (2007) 179(12):8274–9. doi: 10.4049/jimmunol.179.12.8274
355. Onishi RM, Gaffen SL. Interleukin-17 and its Target Genes: Mechanisms of Interleukin-17 Function in Disease. Immunology (2010) 129(3):311–21. doi: 10.1111/j.1365-2567.2009.03240.x
356. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of Interleukin 17 Receptor Signaling for Lung CXC Chemokine and Granulocyte Colony-Stimulating Factor Expression , Neutrophil Recruitment , and Host Defense. J Exp Med (2001) 194(4):519–27. doi: 10.1084/jem.194.4.519
357. Griffin GK, Newton G, Tarrio ML, Maganto-Garcia E, Azcutia V, Alcaide P, et al. IL-17 and Tnfα Sustain Neutrophil Recruitment During Inflammation Through Synergistic Effects on Endothelial Activation. J Immunol (2012) 188(12):6287–99. doi: 10.4049/jimmunol.1200385
358. Eder C. Mechanisms of Interleukin-1β Release. Immunobiology (2009) 214(7):543–53. doi: 10.1016/j.imbio.2008.11.007
359. Gabay C, Lamacchia C, Palmer G. IL-1 Pathways in Inflammation and Human Diseases. Nat Rev Rheumatol (2010) 6(4):232–41. doi: 10.1038/nrrheum.2010.4
360. Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The Interleukin (IL)-1 Cytokine Family - Balance Between Agonists and Antagonists in Inflammatory Diseases. Cytokine (2015) 76(1):25–37. doi: 10.1016/j.cyto.2015.06.017
361. Arend WP, Malyak M, Guthridge CJ. Interleukin-1 Receptor Antagonist: Role in Biology. Annu Rev Immunol (1998) 16:27–55. doi: 10.1146/annurev.immunol.16.1.27
362. Colotta F, Dower SK, Sims JE, Mantovani A. The Type II ‘ Decoy ‘ Receptor : A Novel Regulatory Pathway for Interleukin 1. Immunol Today (1994) 15(12):562–6. doi: 10.1016/0167-5699(94)90217-8
363. Burger D, Chicheportiche R, Giri JG, Dayer J. The Inhibitory Activity of Human Interleukin-1 Receptor Antagonist is Enhanced by Type II Interleukin-1 Soluble Receptor and Hindered by Type I Interleukin-1 Soluble Receptor. J Clin Invest (1995) 96:38–41. doi: 10.1172/JCI118045
364. Martin P, Palmer G, Vigne S, Lamacchia C, Rodriguez E, Talabot-Ayer D, et al. Mouse Neutrophils Express the Decoy Type 2 Interleukin-1 Receptor (IL-1r2) Constitutively and in Acute Inflammatory Conditions. J Leukoc Biol (2013) 94(4):791–802. doi: 10.1189/jlb.0113035
365. Sambo P, Fadlon EJ, Sironi M, Matteucci C, Introna M, Mantovani A, et al. Reactive Oxygen Intermediates Cause Rapid Release of the Interleukin-1 Decoy Receptor From Human Myelomonocytic Cells. Blood (1996) 87(5):1682–6. doi: 10.1182/blood.V87.5.1682.1682
366. Colotta BF, Orlando S, Fadlon EJ, Sozzani S, Matteucci C, Mantovani A, et al. Chemoattractants Induce Rapid Release of the Interleukin 1 Type II Decoy Receptor in Human Polymorphonuclear Cells. J Exp Med (1995) 181:2181–8. doi: 10.1084/jem.181.6.2181
367. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic Interleukin-1 Exposure Drives Haematopoietic Stem Cells Towards Precocious Myeloid Differentiation at the Expense of Self-Renewal. Nat Cell Biol (2016) 18(6):607–18. doi: 10.1038/ncb3346
368. Ueda Y, Cain DW, Kuraoka M, Kondo M, Kelsoe G. IL-1r Type I-Dependent Hemopoietic Stem Cell Proliferation Is Necessary for Inflammatory Granulopoiesis and Reactive Neutrophilia. J Immunol (2009) 182:6477–84. doi: 10.4049/jimmunol.0803961
369. Gallicchio VS, Watts TD, DellaPuca R. Synergistic Action of Recombinant-Derived Murine Interleukin-1 on the Augmentation of Colony Stimulating Activity on Murine Granulocyte-Macrophage Hematopoietic Stem Cells In Vitro. Exp Cell Biol (1987) 55(2):83–92. doi: 10.1159/000163402
370. Opdenakker G, Fibbe WE, Van Damme J. The Molecular Basis of Leukocytosis. Immunol Today (1998) 19(4):182–9. doi: 10.1016/S0167-5699(97)01243-7
371. Walker F, Zhang H, Matthews V, Weinstock J, Nice EC, Ernst M, et al. IL6 / Sil6r Complex Contributes to Emergency Granulopoietic Responses in G-CSF- and GM-CSF-Deficient Mice. Blood (2008) 111(8):3978–85. doi: 10.1182/blood-2007-10-119636
372. Dinarello CA. Biologic Basis for Interleukin-1 in Disease. Blood (1996) 15(87):2095–147. doi: 10.1182/blood.V87.6.2095.bloodjournal8762095
373. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, et al. IL-17-Producing Gamma Delta T Cells and Neutrophils Conspire to Promote Breast Cancer Metastasis. Nature (2015) 522:345–8. doi: 10.1038/nature14282
374. Biondo C, Mancuso G, Midiri A, Signorino G, Domina M, Cariccio VL, et al. The Interleukin-1b/CXCL1/2/Neutrophil Axis Mediates Host Protection Against Group B Streptococcal Infection. Infect Immun (2014) 82(11):4508–17. doi: 10.1128/IAI.02104-14
375. Strieter RM, Kunkel SL, Showell HJ, Remick DG, Phan SH, Ward PA, et al. Endothelial Cell Gene Expression of a Neutrophil Chemotactic Factor by TNF-Alpha, LPS, and IL-1 Beta. Sci (80- ) (1989) 243(4897):1467–9. doi: 10.1126/science.2648570
376. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil Extracellular Trap Formation Is Associated With IL-1b and Autophagy-Related Signaling in Gout. PloS One (2011) 6(12):e29318. doi: 10.1371/journal.pone.0029318
377. Prince LR, Allen L, Jones EC, Hellewell PG, Dower SK, Whyte MKB, et al. The Role of Interleukin-1b in Direct and Toll-Like Receptor 4-Mediated Neutrophil Activation and Survival. Am J Pathol (2004) 165(5):1819–26. doi: 10.1016/S0002-9440(10)63437-2
378. Yagisawa M, Yuo A, Kitagawa S, Yazaki Y, Togawa A, Takaku F. Stimulation and Priming of Human Neutrophils by IL-1 Alpha and IL-1 Beta: Complete Inhibition by IL-1 Receptor Antagonist and No Interaction With Other Cytokines. Exp Hematol (1995) 23(7):603–8.
379. Put K, Vandenhaute J, Avau A, van Nieuwenhuijze A, Brisse E, Dierckx T, et al. Inflammatory Gene Expression Profile and Defective Interferon-γ and Granzyme K in Natural Killer Cells From Systemic Juvenile Idiopathic Arthritis Patients. Arthritis Rheumatol (2017) 69(1):213–24. doi: 10.1002/art.39933
380. Prahalad S, Martins TB, Tebo AE, Whiting A, Clifford B, Zeft AS, et al. Elevated Serum Levels of Soluble CD154 in Children With Juvenile Idiopathic Arthritis. Pediatr Rheumatol (2008) 6:8. doi: 10.1186/1546-0096-6-8
381. Yilmaz M, Kendirli SG, Altintas D, Bingöl G, Antmen B. Cytokine Levels in Serum of Patients With Juvenile Rheumatoid Arthritis. Clin Rheumatol (2001) 20(1):30–5. doi: 10.1007/s100670170100
382. De Benedetti F, Pignatti P, Massa M, Sartirana P, Ravelli A, Martini A. Circulating Levels of Interleukin 1 Beta and of Interleukin 1 Receptor Antagonist in Systemic Juvenile Chronic Arthritis. Clin Exp Rheumatol (1995) 13(6):779–84.
383. Ogilvie EM, Khan A, Hubank M, Kellam P, Woo P. Specific Gene Expression Profiles in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheum (2007) 56(6):1954–65. doi: 10.1002/art.22644
384. Barnes MG, Grom AA, Thompson SD, Griffin TA, Pavlidis P, Itert L, et al. Subtype-Specific Peripheral Blood Gene Expression Profiles in Recent-Onset Juvenile Idiopathic Arthritis. Arthritis Rheum (2009) 60(7):2102–12. doi: 10.1002/art.24601
385. Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, et al. Blood Leukocyte Microarrays to Diagnose Systemic Onset Juvenile Idiopathic Arthritis and Follow the Response to IL-1 Blockade. J Exp Med (2007) 204(9):2131–44. doi: 10.1084/jem.20070070
386. Macaubas C, Nguyen KD, Peck A, Buckingham J, Deshpande C, Wong E, et al. Alternative Activation in Systemic Juvenile Idiopathic Arthritis Monocytes. Clin Immunol (2012) 142(3):362–72. doi: 10.1016/j.clim.2011.12.008
387. Giancane G, Minoia F, Davì S, Bracciolini G, Consolaro A, Ravelli A. IL-1 Inhibition in Systemic Juvenile Idiopathic Arthritis. Front Pharmacol (2016) 7:467. doi: 10.3389/fphar.2016.00467
388. Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful Treatment of Severe Paediatric Rheumatic Disease-Associated Macrophage Activation Syndrome With Interleukin-1 Inhibition Following Conventional Immunosuppressive Therapy: Case Series With 12 Patients. Rheumatol (2011) 50(2):417–9. doi: 10.1093/rheumatology/keq218
389. Vastert SJ, Jamilloux Y, Quartier P, Ohlman S, Franck-larsson K, Koskinen LO, et al. Anakinra in Children and Adults With Still’s Disease. Rheumatology (2019) 58(6):vi9–vi22. doi: 10.1093/rheumatology/kez350
390. Nirmala N, Brachat A, Feist E, Blank N, Specker C, Witt M, et al. Gene-Expression Analysis of Adult-Onset Still ‘ s Disease and Systemic Juvenile Idiopathic Arthritis is Consistent With a Continuum of a Single Disease Entity. Pediatr Rheumatol (2015) 13:50. doi: 10.1186/s12969-015-0047-3
391. Tanaka T, Kishimoto T. Targeting Interleukin-6 : All the Way to Treat Autoimmune and Inflamma- Tory Diseases. Int J Biol Sci (2012) 8(9):1227–36. doi: 10.7150/ijbs.4666
392. Hunter CA, Jones SA. IL-6 as a Keystone Cytokine in Health and Disease. Nat Immunol (2015) 16(5):448–58. doi: 10.1038/ni.3153
393. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Tadamitsu K, et al. Impaired Immune and Acute-Phase Respones in Interleukin-6-Deficient Mice. Nature (1994) 368:815–8. doi: 10.1038/368339a0
394. Maeda K, Baba Y, Nagai Y, Miyazaki K, Malykhin A, Nakamura K, et al. IL-6 Blocks a Discrete Early Step in Lymphopoiesis. Blood (2005) 106(3):879–85. doi: 10.1182/blood-2005-02-0456
395. Biswas BP, Delfanti F, Bernasconi S, Mengozzi M, Cota M, Polentarutti N, et al. Interleukin-6 Induces Monocyte Chemotactic Protein-1 in Peripheral Blood Mononuclear Cells and in the U937 Cell Line. Blood (1998) 91(1):258–65. doi: 10.1182/blood.V91.1.258
396. Pojda Z, Tsuboi A. In Vivo Effects of Human Recombinant Interleukin 6 on Hemopoietic Stem and Progenitor Cells and Circulating Blood Cells in Normal Mice. Exp Hematol (1990) 18(9):1034–7.
397. Suwa T, Hogg JC, English D, Van Eeden SF. Interleukin-6 Induces Demargination of Intravascular Neutrophils and Shortens Their Transit in Marrow. Am J Physiol Hear Circ Physiol (2000) 279:2954–60. doi: 10.1152/ajpheart.2000.279.6.H2954
398. Ulich TR, del CJ, Guo K. In Vivo Hematologic Effects of Recombinant Interleukin-6 on Hematopoiesis and Circulating Numbers of RBCs and WBCs. Blood (1989) 73(1):108–10. doi: 10.1182/blood.V73.1.108.bloodjournal731108
399. Hashizume M, Higuchi Y, Uchiyama Y, Mihara M. IL-6 Plays an Essential Role in Neutrophilia Under Inflammation. Cytokine (2011) 54(1):92–9. doi: 10.1016/j.cyto.2011.01.007
400. Suwa T, Hogg JC, Klut ME, Hards J, Van Eeden SF. Interleukin-6 Changes Deformability of Neutrophils and Induces Their Sequestration in the Lung. Am J Respir Crit Care Med (2001) 163:970–6. doi: 10.1164/ajrccm.163.4.2005132
401. Reynaud D, Pietras E, Barry-holson K, Mir A, Binnewies M, Jeanne M, et al. IL-6 Controls Leukemic Multipotent Progenitor Cell Fate and Contributes to Chronic Myelogenous Leukemia Development. Cancer Cell (2012) 20(5):661–73. doi: 10.1016/j.ccr.2011.10.012
402. Romani L, Mencacci A, Cenci E, Spaccapelo R, Toniatti C, Puccetti P, et al. Impaired Neutrophil Response and CD4+ T Helper Cell 1 Development in Interleukin 6-Deficient Mice Infected With Candida Albicans. J Exp Med (1996) 183:1345–55. doi: 10.1084/jem.183.4.1345
403. Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, et al. IL-6 Receptor Inhibition With Tocilizumab Improves Treatment Outcomes in Patients With Rheumatoid Arthritis Refractory to Anti-Tumour Necrosis Factor Biologicals : Results From a 24-Week Multicentre Randomised Placebo-Controlled Trial. Ann Rheum Dis (2008) 67:1516–23. doi: 10.1136/ard.2008.092932
404. Genovese MC, Mckay JD, Nasonov EL, Mysler EF, Silva NA, Alecock E, et al. Interleukin-6 Receptor Inhibition With Tocilizumab Reduces Disease Activity in Rheumatoid Arthritis With Inadequate Response to Disease-Modifying Antirheumatic Drugs The Tocilizumab in Combination With Traditional Disease-Modifying Antirheumatic Drug. Arthritis Rheum (2008) 58(10):2968–80. doi: 10.1002/art.23940
405. Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, et al. Effect of Interleukin-6 Receptor Inhibition With Tocilizumab in Patients With Rheumatoid Arthritis (OPTION Study): A Double-Blind, Placebo-Controlled, Randomised Trial. Lancet (2008) 371(9617):987–97. doi: 10.1016/S0140-6736(08)60453-5
406. Maini RN, Taylor PC, Szechinski J, Pavelka K, Bro J, Balint G, et al. Double-Blind Randomized Controlled Clinical Trial of the Interleukin-6 Receptor Antagonist , Tocilizumab , in European Patients With Rheumatoid Arthritis Who Had an Incomplete Response to Methotrexate. Arthritis Rheum (2006) 54(9):2817–29. doi: 10.1002/art.22033
407. Jones G, Sebba A, Gu J, Lowenstein MB, Calvo A, Siri DA, et al. Comparison of Tocilizumab Monotherapy Versus Methotrexate Monotherapy in Patients With Moderate to Severe Rheumatoid Arthritis : The AMBITION Study. Ann Rheum Dis (2010) 69:88–96. doi: 10.1136/ard.2008.105197
408. Nakamura I, Omata Y, Naito M, Ito K. Blockade of Interleukin 6 Signaling Induces Marked Neutropenia in Patients With Rheumatoid Arthritis. J Rheumatol (2009) 36(2):459. doi: 10.3899/jrheum.080930
409. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of Interleukin (IL)-6-Type Cytokine Signalling and its Regulation. Biochemistry (2003) 374(Pt 1):1–20. doi: 10.1042/bj20030407
410. Marin V, Montero-Julian F, Grès S, Bongrand P, Farnarier C, Kaplanski G. Chemotactic Agents Induce IL-6ra Shedding From Polymorphonuclear Cells : Involvement of a Metalloproteinase of the TNFa -Converting Enzyme (TACE) Type. Eur J Immunol (2002) 32:2965–70. doi: 10.1002/1521-4141(2002010)32:10<2965::AID-IMMU2965>3.0.CO;2-V
411. Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS, et al. Transgenic Blockade of Interleukin 6 Transsignaling Abrogates Inflammation. Blood (2008) 111(3):1021–8. doi: 10.1182/blood-2007-07-102137
412. Hurst SM, Wilkinson TS, Mcloughlin RM, Jones S, Horiuchi S, Yamamoto N, et al. IL-6 and Its Soluble Receptor Orchestrate a Temporal Switch in the Pattern of Leukocyte Recruitment Seen During Acute Inflammation. Immunity (2001) 14:705–14. doi: 10.1016/S1074-7613(01)00151-0
413. Marin V, Montero-julian FA, Grès S, Bongrand P, Farnarier C, Boulay V, et al. The IL-6-Soluble IL-6r α Autocrine Loop of Endothelial Activation as an Intermediate Between Acute and Chronic Inflammation: An Experimental Model Involving Thrombin. J Immunol (2001) 167:3435–42. doi: 10.4049/jimmunol.167.6.3435
414. Gabay C. Interleukin-6 and Chronic Inflammation. Arthritis Res Ther (2006) 8(2):S3. doi: 10.1186/ar1917
415. Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, et al. Role of IL-6 and Its Soluble Receptor in Induction of Chemokines and Leukocyte Recruitment. Immunity (1997) 6:315–25. doi: 10.1016/S1074-7613(00)80334-9
416. Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei X, et al. IL-6 Is an Antiinflammatory Cytokine Required for Controlling Local or Systemic Acute Inflammatory Responses. J Clin Invest (1998) 101(2):311–20. doi: 10.1172/JCI1368
417. Atreya R, Mudter J, Finotto S, Müllberg J, Jostock T, Wirtz S, et al. Blockade of Interleukin 6 Trans Signaling Suppresses T-Cell Resistance Against Apoptosis in Chronic Intestinal Inflammation : Evidence in Crohn Disease and Experimental Colitis In Vivo. Nat Med (2000) 6(5):583–8. doi: 10.1038/75068
418. Alonzi BT, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, et al. Interleukin 6 Is Required for the Development of Collagen-Induced Arthritis. J Exp Med (1998) 187(4):461–8. doi: 10.1084/jem.187.4.461
419. Yamamoto M, Yoshizaki K, Ito H, Alerts E. IL-6 Is Required for the Development of Th1 Cell-Mediated Murine Colitis. J Immunol (2000) 164:4878–82. doi: 10.4049/jimmunol.164.9.4878
420. Tanaka T, Narazaki M, Kishimoto T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol (2014) 6(10):a016295. doi: 10.1101/cshperspect.a016295
421. Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, et al. Essential Role of IL-6 in Protection Against H1N1 Influenza Virus by Promoting Neutrophil Survival in the Lung. Nature (2012) 5(3):258–66. doi: 10.1038/mi.2012.2
422. Wright HL, Cross AL, Edwards SW, Moots RJ. Effects of IL-6 and IL-6 Blockade on Neutrophil Function In Vitro and In Vivo. Rheumatology (2014) 53:1321–31. doi: 10.1093/rheumatology/keu035
423. Afford SC, Pongracz J, Stockley RA, Crocker J, Burnett D, Cells U. The Induction by Human Interleukin-6 of Apoptosis in the Promonocytic Cell Line U937 and Human Neutrophils. J Biol Chem (1992) 267(30):21612–6. doi: 10.1016/S0021-9258(19)36655-4
424. Biffl WL, Moore EE, Moore FA, Banett CC Jr. Interleukin-6 Suppression of Neutrophil Apoptosis is Neutrophil Concentration Dependent. J Leukoc Biol (1995) 58(5):582–4. doi: 10.1002/jlb.58.5.582
425. Mcnamee JP, Bellier PV, Kutzner BC, Wilkins RC. Effect of Pro-Inflammatory Cytokines on Spontaneous Apoptosis in Leukocyte Sub-Sets Within a Whole Blood Culture. Cytokine (2005) 31:161–7. doi: 10.1016/j.cyto.2005.05.001
426. Ottonello L, Frumento G, Arduino N, Bertolotto M, Dapino P, Mancini M, et al. Differential Regulation of Spontaneous and Immune Complex- Induced Neutrophil Apoptosis by Proinflammatory Cytokines . Role of Oxidants , Bax and Caspase-3. J Leukoc Biol (2002) 72:125–32.
427. Wilkinson AN, Gartlan KH, Kelly G, Samson LD, Olver SD, Avery J, et al. Granulocytes Are Unresponsive to IL-6 Due to an Absence of Gp130. J Immunol (2018) 200(10):3547–55. doi: 10.4049/jimmunol.1701191
428. Calzetti F, Tamassia N, Arruda-Silva F, Gasperini S, Cassatella MA. The Importance of Being “Pure” Neutrophils. J Allergy Clin Immunol (2017) 139(1):352–355.e6. doi: 10.1016/j.jaci.2016.06.025
429. Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct Cytokine Profiles of Systemic-Onset Juvenile Idiopathic Arthritis-Associated Macrophage Activation Syndrome With Particular Emphasis on the Role of Interleukin-18 in its Pathogenesis. Rheumatology (2010) 49:1645–53. doi: 10.1093/rheumatology/keq133
430. Imbrechts M, Avau A, Vandenhaute J, Malengier-Devlies B, Put K, Mitera T, et al. Insufficient IL-10 Production as a Mechanism Underlying the Pathogenesis of Systemic Juvenile Idiopathic Arthritis. J Immunol (2018) 201(9):2654–63. doi: 10.4049/jimmunol.1800468
431. Lotito APN, Campa A, Silva CAA, Kiss MHB, Mello SBV. Interleukin 18 as a Marker of Disease Activity and Severity in Patients With Juvenile Idiopathic Arthritis. J Rheumatol (2007) 34(4):823–30.
432. De Benedetti F, Massa M, Robbioni P, Ravelli A, Burgio GR, Martini A. Correlation of Serum Interleukin-6 Levels With Joint Involement and Thrombocytosis in Systemic Juvenile Rheumatoid Arthritis. Arthritis Rheum (1991) 34(9):1158–63. doi: 10.1002/art.1780340912
433. Shimizu M, Yachie A. Compensated Inflammation in Systemic Juvenile Idiopathic Arthritis : Role of Alternatively Activated Macrophages. Cytokine (2012) 60(1):226–32. doi: 10.1016/j.cyto.2012.05.003
434. Lepore L, Pennesi M, Saletta S, Perticarari S, Presani G, Prodan M. Study of IL-2, IL-6, TNF Alpha, IFN Gamma and Beta in the Serum and Synovial Fluid of Patients With Juvenile Chronic Arthritis. Clin Exp Rheumatol (1994) 12(5):561–32.
435. Shimizu M, Nakagishi Y, Kasai K, Yamasaki Y, Miyoshi M, Takei S. Tocilizumab Masks the Clinical Symptoms of Systemic Juvenile Idiopathic Arthritis-Associated Macrophage Activation Syndrome : The Diagnostic Significance of Interleukin-18 and Interleukin-6. Cytokine (2012) 58(2):287–94. doi: 10.1016/j.cyto.2012.02.006
436. Rooney M, David J, Symons J, Di Giovine F, Varsani H, Woo P. Inflammatory Cytokine Responses in Juvenile Chronic Arthritis. Br J Rheumatol (1995) 34(5):454–60. doi: 10.1093/rheumatology/34.5.454
437. Shimizu M, Nakagishi Y, Inoue N, Mizuta M, Ko G, Saikawa Y, et al. Interleukin-18 for Predicting the Development of Macrophage Activation Syndrome in Systemic Juvenile Idiopathic Arthritis. Clin Immunol (2015) 160(2):277–81. doi: 10.1016/j.clim.2015.06.005
438. De Benedetti F, Martini A. Targeting the Interleukin-6 Receptor : A New Treatment for Systemic Juvenile Idiopathic Arthritis? Arthritis Rheum (2005) 52(3):687–93. doi: 10.1002/art.20946
439. De Benedetti F, Meazza C, Martini A. Role of Interleukin-6 in Growth Failure : An Animal Model Role of Interleukin-6 in Growth Failure : An Animal Model. Horm Res (2002) 58:24–7. doi: 10.1159/000064757
440. Avau A, Put K, Wouters CH, Matthys P. Cytokine Balance and Cytokine-Driven Natural Killer Cell Dysfunction in Systemic Juvenile Idiopathic Arthritis. Cytokine Growth Factor Rev (2015) 26(1):35–45. doi: 10.1016/j.cytogfr.2014.05.005
441. Omoyinmi E, Hamaoui R, Bryant A, Jiang MC, Athigapanich T, Eleftheriou D, et al. Mitochondrial and Oxidative Stress Genes are Differentially Expressed in Neutrophils of sJIA Patients Treated With Tocilizumab : A Pilot Microarray Study. Pediatr Rheumatol (2016) 14(1):7. doi: 10.1186/s12969-016-0067-7
442. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 Binding Protein. Front Immunol (2013) 4:289. doi: 10.3389/fimmu.2013.00289
443. Novick D, Kim S, Kaplanski G, Dinarello CA. Interleukin-18 , More Than a Th1 Cytokine. Semin Immunol (2013) 25(6):439–48. doi: 10.1016/j.smim.2013.10.014
444. Leung BP, Culshaw S, Gracie JA, Canetti CA, Campbell C, Liew FY, et al. A Role for IL-18 in Neutrophil Activation. J Immunol (2001) 167(5):2879–86. doi: 10.4049/jimmunol.167.5.2879
445. Elbim C, Dang PMC, Pedruzzi E, El Benna J, Bichat CHX. Interleukin-18 Primes the Oxidative Burst of Neutrophils in Response to Formyl-Peptides : Role of Cytochrome B558 Translocation and N-Formyl Peptide Receptor Endocytosis. Clin Diagn Lab Immunol (2005) 12(3):436–46. doi: 10.1128/CDLI.12.3.436-446.2005
446. Fortin CF, Ear T, Mcdonald PP. Autocrine Role of Endogenous Interleukin-18 on Inflammatory Cytokine Generation by Human Neutrophils. FASEB J (2009) 23(1):194–203. doi: 10.1096/fj.08-110213
447. Müller AA, Dolowschiak T, Sellin ME, Felmy B, Verbree C. An NK Cell Perforin Response Elicited via IL-18 Controls Mucosal Inflammation Kinetics During Salmonella Gut Infection. PloS Pathog (2016) 12(6):e1005723. doi: 10.1371/journal.ppat.1005723
448. Put K, Avau A, Brisse E, Mitera T, Put S, Proost P, et al. Cytokines in Systemic Juvenile Idiopathic Arthritis and Haemophagocytic Lymphohistiocytosis: Tipping the Balance Between Interleukin-18 and Interferon-γ. Rheumatology (2015) 54(8):1507–17. doi: 10.1093/rheumatology/keu524
449. Chen O, Shan N, Zhu X, Wang Y, Ren P, Wei D, et al. The Imbalance of IL-18 / IL-18BP in Patients With Systemic Juvenile Idiopathic Arthritis. Acta Biochim Biophys Sin (2013) 45(4):339–41. doi: 10.1093/abbs/gmt007
450. de Jager W, Vastert SJ, Beekman JM, Wulffraat NM, Kuis W, Coffer PJ, et al. Defective Phosphorylation of Interleukin-18 Receptor Beta Causes Impaired Natural Killer Cell Function in Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheumatol (2009) 60(9):2782–93. doi: 10.1002/art.24750
451. Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, et al. Open-Label, Multicentre, Dose-Escalating Phase II Clinical Trial on the Safety and Efficacy of Tadekinig Alfa (IL-18BP) in Adult-Onset Still’s Disease. Ann Rheum Dis (2018) 77(6):840–7. doi: 10.1136/annrheumdis-2017-212608
452. Billiau A, Matthys P. Interferon-Gamma : A Historical Perspective. Cytokine Growth Factor Rev (2009) 20:97–113. doi: 10.1016/j.cytogfr.2009.02.004
453. Lin F, Young HA. The Talented Interferon-Gamma. Adv Biosci Biotechnol (2013) 4:6–13. doi: 10.4236/abb.2013.47A3002
454. Kelchtermans H, Billiau A, Matthys P. How Interferon-γ Keeps Autoimmune Diseases in Check. Trends Immunol (2008) 29(10):479–86. doi: 10.1016/j.it.2008.07.002
455. De Bruin AM, Voermans C, Nolte MA. Impact of Interferon-γ on Hematopoiesis. Blood (2014) 124(16):2479–86. doi: 10.1182/blood-2014-04-568451
456. de Bruin AM, Libregts SF, Valkhof M, Boon L, Touw IP, Nolte MA. IFN-Gamma Induces Monopoiesis and Inhibits Neutrophil Development During Inflammation. Blood (2012) 119(6):1543–54. doi: 10.1182/blood-2011-07-367706
457. Snoeck HW, Lardon F, Lenjou M, Nys G, Van Bockstaele DR, Peetermans ME. Interferon-Gamma and Interleukin-4 Reciprocally Regulate the Production of Monocytes/Macrophages and Neutrophils Through a Direct Effect on Committed Monopotential Bone Marrow Progenitor Cells. Eur J Immunol (1993) 23(5):1072–7. doi: 10.1002/eji.1830230514
458. Cruz A, Khader SA, Torrado E, Fraga A, Pearl JE, Pedrosa J, et al. Cutting Edge: IFN- γ Regulates the Induction and Expansion of IL-17-Producing CD4 T Cells During Mycobacterial Infection. J Immunol (2006) 177:1416–20. doi: 10.4049/jimmunol.177.3.1416
459. Desvignes L, Ernst JD. Interferon-Gamma-Responsive Nonhematopoietic Cells Regulate the Immune Response to Mycobacterium Tuberculosis. Immunity (2009) 31(6):974–85. doi: 10.1016/j.immuni.2009.10.007
460. Pearl JE, Saunders B, Ehlers S, Orme IM, Cooper AM. Inflammation and Lymphocyte Activation During Mycobacterial Infection in the Interferon-Gamma-Deficient Mouse. Cell Immunol (2001) 50:43–50. doi: 10.1006/cimm.2001.1819
461. Norose K, Naoi K, Fang H, Yano A. In Vivo Study of Toxoplasmic Parasitemia Using Interferon- γ Deficient Mice : Absolute Cell Number of Leukocytes , Parasite Load and Cell Susceptibility. Parasitol Int (2008) 57(4):447–53. doi: 10.1016/j.parint.2008.05.007
462. Ellison MA, Gearheart CM, Porter CC, Ambruso DR. IFN-γ Alters the Expression of Diverse Immunity Related Genes in a Cell Culture Model Designed to Represent Maturing Neutrophils. PloS One (2017) 12(10):e0185956. doi: 10.1371/journal.pone.0185956
463. Ellis TN, Beaman BL. Interferon-γ Activation of Polymorphonuclear Neutrophil Function. Immunology (2004) 112(1):2–12. doi: 10.1111/j.1365-2567.2004.01849.x
464. Marchi LF, Ignacchiti MDC, Mantovani B. In Vitro Activation of Mouse Neutrophils by Recombinant Human Interferon-Gamma : Increased Phagocytosis and Release of Reactive Oxygen Species and Pro-In Fl Ammatory Cytokines. Int Immunopharmacol (2014) 18(2):228–35. doi: 10.1016/j.intimp.2013.12.010
465. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon- Y : An Overview of Signals , Mechanisms and Functions. J Leukoc Biol (2004) 75:163–89. doi: 10.1189/jlb.0603252
466. Green DS, Young HA, Valencia JC. Current Prospects of Type II Interferon-G Signaling and Autoimmunity. J Biol Chem (2017) 292(9):13925–33. doi: 10.1074/jbc.R116.774745
467. Hu X, Ivashiv LB. Cross-Regulation of Signaling and Immune Responses by IFN-γ and STAT1. Immunity (2010) 31(4):539–50. doi: 10.1016/j.immuni.2009.09.002
468. de Kleijn S, Langereis JD, Leentjens J, Kox M, Netea MG, Koenderman L, et al. IFN-γ-Stimulated Neutrophils Suppress Lymphocyte Proliferation Through Expression of PD-L1. PloS One (2013) 8(8):e72249. doi: 10.1371/journal.pone.0072249
469. Sikora KA, Fall N, Thornton S, Grom AA. The Limited Role of Interferon-γ in Systemic Juvenile Idiopathic Arthritis Cannot Be Explained by Cellular Hyporesponsiveness. Arthritis Rheum (2012) 64(11):3799–808. doi: 10.1002/art.34604
470. Cambier S, Metzemaekers M, Carvalho AC, Nooyens A, Jacobs C, Vanderbeke L, et al. Atypical Response to Bacterial Co-Infection and Persistent Neutrophilic Broncho-Alveolar Inflammation Distinguish Critical COVID-19 From Influenza. JCI Insight (2021) e155055. doi: 10.1172/jci.insight.155055
Keywords: neutrophil, emergency granulopoiesis, inflammation, systemic juvenile idiopathic arthritis, left shift, cytokines
Citation: Malengier-Devlies B, Metzemaekers M, Wouters C, Proost P and Matthys P (2021) Neutrophil Homeostasis and Emergency Granulopoiesis: The Example of Systemic Juvenile Idiopathic Arthritis. Front. Immunol. 12:766620. doi: 10.3389/fimmu.2021.766620
Received: 29 August 2021; Accepted: 23 November 2021;
Published: 13 December 2021.
Edited by:
Dirk Holzinger, Essen University Hospital, GermanyReviewed by:
Claudia Bracaglia, IRCCS Ospedale Pediatrico Bambino Gesù (IRCCS), ItalySeth Lucian Masters, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright © 2021 Malengier-Devlies, Metzemaekers, Wouters, Proost and Matthys. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bert Malengier-Devlies, bert.malengier@kuleuven.be; Patrick Matthys, patrick.matthys@kuleuven.be