Microglia in Alzheimer’s disease: pathogenesis, mechanisms, and therapeutic potentials
 Jifei Miao
Jifei Miao Haixia Ma3
Haixia Ma3  Jiao Lan
Jiao Lan- 1Shenzhen Bao’an Traditional Chinese Medicine Hospital, Shenzhen, China
- 2School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen, China
- 3Shenzhen Bao’an Traditional Chinese Medicine Hospital, Guangzhou University of Chinese Medicine, Shenzhen, China
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by protein aggregation in the brain. Recent studies have revealed the critical role of microglia in AD pathogenesis. This review provides a comprehensive summary of the current understanding of microglial involvement in AD, focusing on genetic determinants, phenotypic state, phagocytic capacity, neuroinflammatory response, and impact on synaptic plasticity and neuronal regulation. Furthermore, recent developments in drug discovery targeting microglia in AD are reviewed, highlighting potential avenues for therapeutic intervention. This review emphasizes the essential role of microglia in AD and provides insights into potential treatments.
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline, memory loss, and changes in behavior and mood. The accumulation of Aβ and tau proteins in the brain is thought to play a significant role in AD pathogenesis. Microglia plays a multifaceted role in AD, contributing to inflammation, phagocytosis, and neurodegeneration. In response to Aβ plaque accumulation, microglia become activated and produce proinflammatory cytokines, leading to a chronic state of neuroinflammation (Hansen et al., 2018). This inflammation has been shown to worsen neurodegeneration and contribute to the progression of AD (Leng and Edison, 2021). Furthermore, as AD progresses, microglia’s ability to phagocytose Aβ plaques decreases, leading to their accumulation in the brain and further AD progression (Hansen et al., 2018). Additionally, microglia contribute to neurodegeneration in AD through the release of toxic substances and regulation of synaptic function (Hong et al., 2016). Understanding the role of microglia in AD is crucial for developing effective treatments. This review provides a comprehensive summary of microglial involvement in AD, including the relevant mutated genes, microglial subtypes, phagocytic function, inflammatory response, and neurodegeneration. It also discusses recent developments in therapy development targeting microglia in AD. Further research is necessary to fully understand the mechanisms by which microglia contribute to AD and to develop new strategies for the treatment of this debilitating disease.
2. Mutant genes in microglia associated with AD
Emerging evidence suggests that mutations in genes associated with microglia significantly impact the development of AD. Microglia play a critical role in monitoring for signs of damage or infection in the brain and responding appropriately. However, mutations in microglia-associated genes can lead to altered microglial functions, including cytokine secretion and phagocytic activity, resulting in increased inflammation in the brain and contributing to AD progression. Moreover, microglial dysfunction has been linked to the clearance of Aβ peptides, key contributors to AD pathology. Mutations in microglia-associated genes may impair this clearance process, leading to the accumulation of Aβ peptides and further cognitive decline. Furthermore, changes in gene expression and intracellular signaling pathways have been linked to mutations in microglia-associated genes, further impairing microglial function and contributing to AD progression. The relationship between mutations in microglia-associated genes and AD underscores the critical role that microglia play in maintaining brain health and protecting against damage and disease. To fully understand the mechanisms of AD onset and progression, it is essential to continue researching these mutated genes and their effects on microglial function. This review provides a comprehensive overview of the current understanding of the relationship between mutations in microglia-associated genes and AD, highlighting the importance of microglial function in the maintenance of brain health and the potential for microglial-targeted therapies.
2.1. APOE
The APOE gene has three common alleles, ε2, ε3, and ε4, that result from single-nucleotide polymorphisms (Sims et al., 2020). The ε4 allele is a strong risk factor for late-onset Alzheimer’s Disease (LOAD), with individuals carrying one APOE ε4 allele having a 3–4-fold higher risk for AD, while those with two ε4 alleles have a 10–15-fold higher risk (Corder et al., 1993; Saunders et al., 1993; Sims et al., 2020). This allele is highly enriched in AD patients and is expressed in both the brain and liver, playing a role in lipoprotein transportation and modulating the inflammatory response (Goldstein and Brown, 2015; Flowers and Rebeck, 2020). The contribution of APOE4 to AD pathogenesis has been attributed primarily to its induction of early and abundant amyloid pathology. However, recent evidence suggests that APOE4 can also independently worsen tau pathology and neurodegeneration (Yamazaki et al., 2019; Koutsodendris et al., 2022). Clinical studies have found a correlation between APOE ε4 and more severe tau pathology, neurodegeneration, and memory impairment in AD patients (Filippini et al., 2009; Therriault et al., 2020; Wang Y.-T. T. et al., 2021; Weigand et al., 2021). APOE ε4 allele is strongly associated with Aβ pathology in human AD brains, with homozygous carriers having the highest Aβ burden (Polvikoski et al., 1995; Premkumar et al., 1996; Bertram et al., 2007; Reiman et al., 2009; Mishra et al., 2018). Reducing APOE levels has been shown to be an attractive strategy for alleviating Aβ pathology in AD patients carrying the APOE ε4 allele. Studies using human iPSC-derived neurons and cerebral organoids have shown that APOE4 exacerbates tau phosphorylation and synapse loss, leading to neurodegeneration (Lin et al., 2018; Wang C. et al., 2018; Zhao et al., 2020a). APOE4 knock-in mice exhibit loss of hilar GABAergic interneurons and memory deficits (Andrews-Zwilling et al., 2010). Depletion of astrocytic APOE4 reduces tau pathology and neurodegeneration in PS19-APOE4 knock-in mice, indicating that reducing APOE4 levels may be an effective strategy for alleviating tau pathology and neurodegeneration in AD therapy (Wang C. et al., 2021). As a potential therapeutic target, various approaches to reducing APOE4 are being explored, including converting it to APOE2 or APOE3 (Safieh et al., 2019; Yamazaki et al., 2019; Williams et al., 2020; McDade et al., 2021). It is noteworthy that the increase in APOE expression in microglia may be protective or detrimental to the disease, and further investigation is needed to determine the stage of the disease when APOE reduction may be effective. This review provides a comprehensive overview of the current understanding of the role of APOE ε4 in AD, highlighting the potential of reducing APOE4 levels as a therapeutic strategy for AD.
2.2. TREM2
TREM2 is a transmembrane receptor that is predominantly expressed in tissue macrophages, including microglia in the brain. It belongs to the immunoglobulin superfamily and is composed of an extracellular IgV domain, a stalk, a single transmembrane helix, and a cytosolic tail (Kober et al., 2016). TREM2 binds to several ligands, including apoptotic cells, phospholipids, glycolipids, lipidated particles, and lipoproteins (Song et al., 2017). Mutations in TREM2 are linked to Nasu-Hakola disease (Paloneva et al., 2002), and heterozygous variants have been associated with an increased risk of developing LOAD (Guerreiro et al., 2013; Jonsson et al., 2013). In AD, TREM2 plays a crucial role in regulating microglial activation. Activated microglia form a barrier that limits the spreading and toxicity of Aβ, and increasing TREM2 expression through overexpression of human TREM2 has been shown to reduce Aβ deposition (Lee et al., 2018). Moreover, agonistic antibodies against TREM2 have been shown to reduce Aβ load and improve behavioral performance (Schlepckow et al., 2020; Ellwanger et al., 2021). TREM2 also helps microglia clear myelin debris and promote remyelination, making it a potential therapeutic target for demyelinating diseases (Plemel et al., 2018; Claes et al., 2021; Zirngibl et al., 2022). The soluble form of TREM2, sTREM2, has been found to be a useful marker of AD pathology and cognitive decline, with higher levels correlating positively with neurodegenerative markers and slower cognitive decline (Henjum et al., 2016; Heslegrave et al., 2016; Suárez-Calvet et al., 2016b; Ewers et al., 2019, 2020; Pascoal et al., 2021). However, the impact of TREM2 cleavage and sTREM2 on AD is not yet clear, as some studies have found a protective effect while others have found a detrimental effect. sTREM2 has been found to bind to Aβ aggregates and impact Aβ pathology, and may activate an independent signaling pathway, presumably by activating the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway (Wu et al., 2015; Zhong et al., 2017, 2019). Investigating the role of TREM2 in microglial cells is crucial to better understand the molecular mechanisms underlying AD and to develop new therapies that target microglia and TREM2.
2.3. CD33
CD33 is predominantly expressed by microglia in the brain and has been investigated for its association with AD susceptibility. Through genome-wide association studies (GWAS), the rs3826656 single nucleotide polymorphism (SNP) and nearby SNP, rs3865444, have been identified as susceptibility factors for LOAD (Hollingworth et al., 2011; Karch et al., 2012). Post-mortem analyses have shown increased expression of CD33 on microglia in AD brains, which correlates with Aβ burden and cognitive decline (Karch et al., 2012; Griciuc et al., 2013). However, higher CD33 expression in AD could also be a response to disease pathology, such as inflammation (Nomura et al., 2001; Ramsborg and Papoutsakis, 2007). Initially, the rs3865444 SNP, located in the CD33 promoter, was thought to modulate CD33 gene expression (Malik et al., 2013). However, further analysis revealed a co-inherited SNP, rs12459419, located in exon 2 of CD33, which impacts mRNA splicing and mediates the expression of two isoforms of CD33: a longer isoform, hCD33M, and a shorter isoform, hCD33m. The rs12459419C allele, co-inherited with the rs3865444C allele, results in hCD33M:hCD33m transcript ratios of 9:1, while the AD-protective rs12459419T allele (i.e., rs3865444A) shifts this ratio to 7:3 (Malik et al., 2013). Cells expressing the rs12459419T allele show decreased levels of hCD33M at the protein level (Bradshaw et al., 2013; Malik et al., 2013). Higher levels of hCD33M expression are associated with decreased phagocytosis in various cells, as demonstrated in primary monocytes and cultured mouse microglia BV2 cells (Bradshaw et al., 2013; Griciuc et al., 2013). Recent studies have shown that hCD33M can decrease the uptake of various cargos, including polystyrene beads, dextran, myelin, and aggregated Aβ1-42 (Bhattacherjee et al., 2019, 2021a). Recent studies have reported a gain-of-function role for hCD33m in microglia, leading to increased phagocytosis, cell migration, and cell proliferation while decreasing cell adhesion (Bhattacherjee et al., 2021b). The two hCD33 isoforms appear to play distinct roles in microglia. Another CD33 SNP, rs2455069, has been proposed to be associated with AD susceptibility, with in silico analysis suggesting that an amino acid switch at position 69 of hCD33 from an arginine to glycine in the rare rs2455069 SNP may enhance the affinity for sialic acid-containing ligands (Tortora et al., 2022). However, further testing is needed to establish this association within a larger cohort.
The investigation of CD33’s association with AD susceptibility and its effects on microglia provides insights into the role of microglia in AD pathology. However, the exact mechanisms by which CD33 isoforms influence microglial function and their impact on AD progression remain to be fully elucidated. Further research is necessary to fully understand the role of CD33 in AD and to develop potential therapeutic strategies targeting microglia and CD33 in AD treatment.
2.4. Other genes
Early research on AD employed cost-effective approaches such as targeted sequencing to identify rare variants in known AD-related genes. In contrast, more recent studies have dedicated substantial resources to whole exome sequencing (WES) or whole genome sequencing (WGS) to uncover rare variants in previously unidentified loci. The AD Sequencing Project (ADSP) conducted a WGS family-based study and a WES case-control study on over 30,000 samples (Beecham et al., 2017), uncovering rare SORL1 loss-of-function variants, TREM2R47H, a common variant in PILRA, and a novel rare variant in long non-coding RNA AC099552.4 (Raghavan et al., 2018; Bis et al., 2020). Gene-level analyses implicated OPRL1 and GAS2L2 in AD, suggesting transcriptional regulation by ZNF655 (Bis et al., 2020). Another investigation identified a novel variant in the NSF gene associated with AD (Fan et al., 2020), while a weighted burden analysis of ADSP’s WES subjects implicated rare variants in TREM2, ABCA7, SORL1, and PSEN1 in AD (Curtis et al., 2020). The PI3K/AKT signaling pathway was found to be significantly associated with TREM2, with PIK3R1, WNT7A, CR1, and EXOC5 likely containing detrimental rare variants and TIAF1 and NDRG2 potentially harboring protective rare variants (Curtis et al., 2020). The largest exome analysis to date corroborated established rare variants in TREM2, SORL1, and ABCA7 loci and identified novel rare variants in the microglial gene ATP8B4 (Holstege, 2020). WGS detected rare variants in non-coding regions, with ABCA1, TMEM132A, and AKAP9 segregating with AD in families and AKAP9 being nominally associated with LOAD risk (Beecham et al., 2018; Vardarajan et al., 2018). Rare variants in CR1, BIN1, FERMT2, and SLC24A4 were also pinpointed, aligning with genome-wide association study (GWAS) findings (Beecham et al., 2018; Vardarajan et al., 2018). A family-based WGS association study identified FNBP1L, SEL1L, LINC00298, PRKCH, C15ORF41, C2CD3, KIF2A, APC, LHX9, NALCN, CTNNA2, SYTL3, and CLSTN2, implicating genes involved in neuroplasticity, synaptic function, and neurodevelopmental pathways, thus broadening the range of AD-related biological pathways (Prokopenko et al., 2021). Collectively, these studies have confirmed and offered novel insights into AD-related biological pathways, emphasizing the significance of WGS in identifying genetic variants in non-coding regions and previously overlooked AD pathogenesis pathways. In this review, we have summarized 37 microglial mutant genes associated with AD (Table 1). Further independent replication of these findings is necessary due to the rarity of some variants.
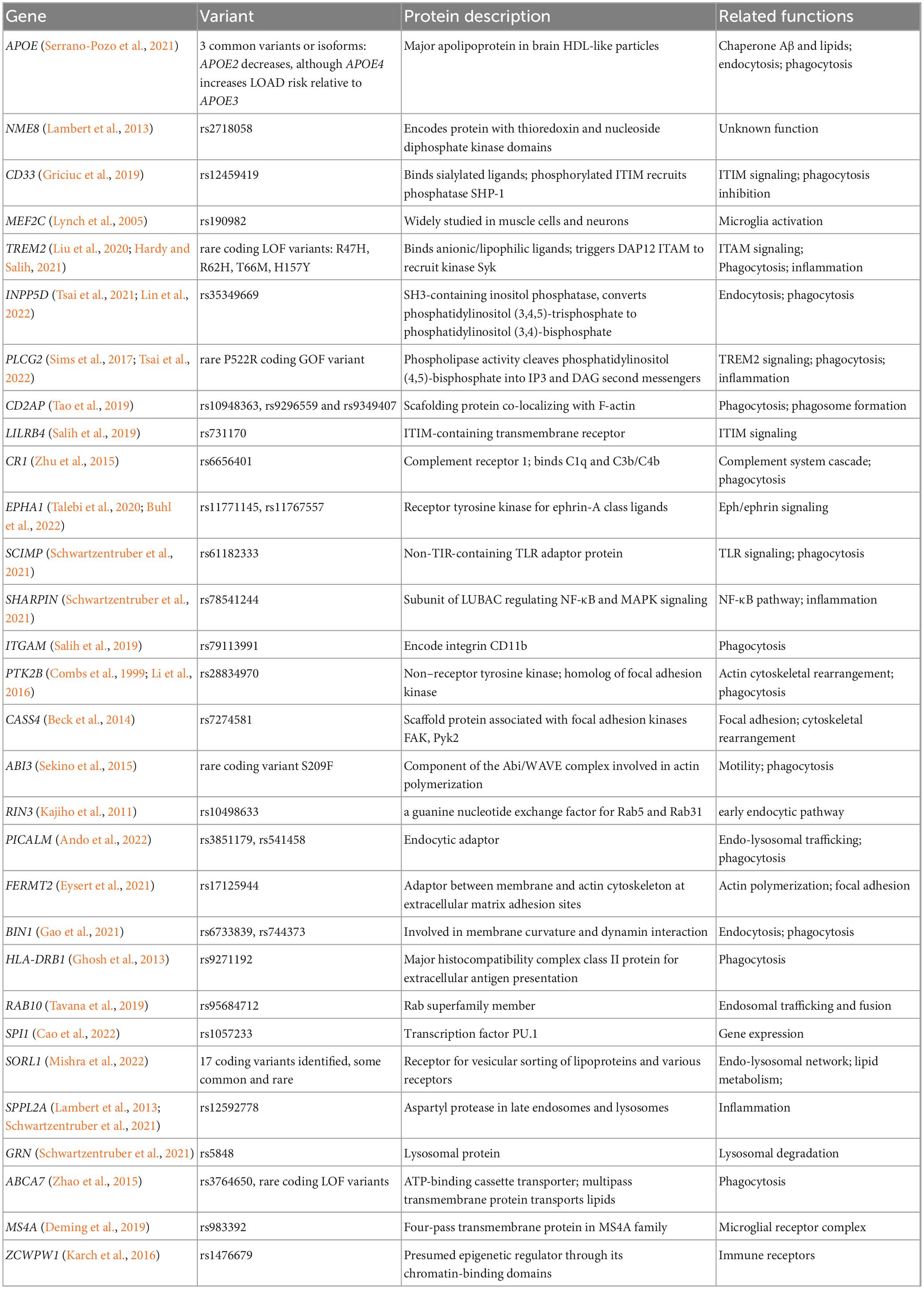
Table 1. Genes in microglia associated with AD.
3. Microglia activation
Microglia exhibit diverse phenotypes in response to environmental cues and display heterogeneity in their activation, which can be broadly classified into two categories: classical (M1) and alternative (M2) (Sica and Mantovani, 2012; Tang and Le, 2016). M1 activation provokes inflammation and neurotoxicity, while M2 activation elicits anti-inflammatory and reparative responses (Tang and Le, 2016; Colonna and Butovsky, 2017). Microglia can adapt their phenotype to different environments, ensuring a protective role (Du et al., 2018). However, recent transcriptomic investigations reveal that microglial activation is multifaceted, with various intermediate phenotypes between M1 and M2 (Correale, 2014). Researchers have also identified a novel subpopulation of microglia associated with neurodegenerative diseases, termed disease-associated microglia (DAM), which localize around Aβ plaques in AD and exhibit a distinct gene expression profile (Keren-Shaul et al., 2017). DAM activation comprises a two-step process involving TREM2-independent and TREM2-dependent mechanisms (Keren-Shaul et al., 2017). The binary classification of microglia as M1 or M2 has been contested, and researchers propose that a continuum of different intermediate phenotypes more accurately reflects microglial activation states. M2 microglia uptake and remove Aβ deposits, protect against AD, and mitigate neurotoxic Aβ aggregation (Song and Suk, 2017). However, uncontrolled microglial activation and aberrant responses to Aβ can harm neurons, resulting in neuroinflammation, synapse loss, and tau pathology (Tang and Le, 2016; Sarlus and Heneka, 2017; Hansen et al., 2018). Microglia continuously transform among phenotypes, and two peaks of microglial activation in AD have been proposed: an early anti-inflammatory peak during the pre-clinical stage and a later pro-inflammatory peak during the clinical stage as the disease advances (Fan et al., 2017). In summary, proper microglial function is crucial in AD pathology, and dysregulation can have detrimental consequences on neurons. Comprehending the dynamic nature of microglial activation and their dual role in AD pathogenesis is vital for developing effective therapies for AD. In this context, further investigation into the signaling pathways involved in microglial activation, such as TLRs, NF-κB, MAPK, PI3K/AKT, and JAK/STAT pathways, is essential (Figure 1). Understanding these pathways’ roles in microglial function can facilitate the development of targeted therapies for AD.
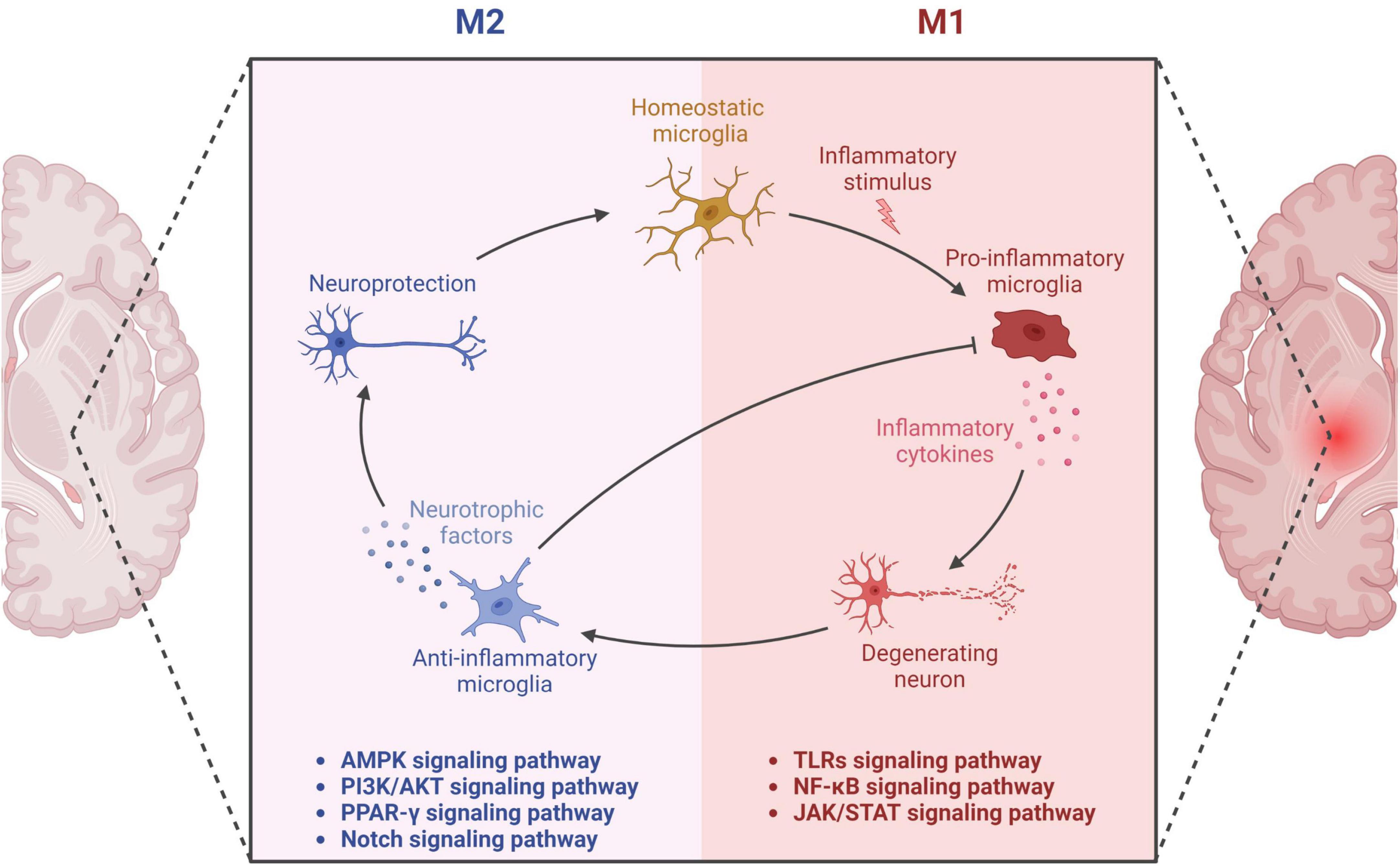
Figure 1. Microglial activation in neurodegeneration and neuroprotection. The diagram showcases the dual functions of microglia through M1 and M2 phenotypes. The M1 microglial phenotype promotes neuroinflammation through the secretion of pro-inflammatory factors, leading to neuronal damage. The related pathways involved in this process encompass Toll-like Receptors (TLRs), Nuclear Factor-kappa B (NF-κB), and the Janus Kinase/Signal Transducers and Activators of Transcription (JAK/STAT) pathway. On the other hand, the M2 phenotype of microglia secretes anti-inflammatory factors, thereby providing neuroprotective effects. This aspect is mediated through various signaling pathways including Adenosine Monophosphate-Activated Protein Kinase (AMPK), Phosphatidylinositol 3-Kinase/Protein Kinase B (PI3K/AKT), Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ), and the Notch pathway. Thus, the diagram delineates the dual role of microglia in neuronal health and disease, underlining the critical balance between the M1 and M2 states.
3.1. Pro-inflammatory (M1) polarization
3.1.1. TLRs signaling pathway
The transmembrane pattern-recognition receptor family, including TLR2 and TLR4, exhibits significant expression in microglia (Cui et al., 2020). TLR4 is noted for its ability to bind lipopolysaccharide (LPS), an interaction that sparks pro-inflammatory signaling pathways and M1 microglial polarization, both of which have been implicated in AD’s neuroinflammatory pathogenesis (Yang Z. et al., 2019). This LPS-TLR4 complex initiates a cascade of inflammatory signaling, beginning with TLR4’s association with myeloid differentiation factor 88 (MyD88) and leading to the autophosphorylation of interleukin-1 receptor-associated kinase (IRAK) (Subhramanyam et al., 2019). This subsequently activated IRAK1 and IRAK4 interact with tumor necrosis factor receptor-associated factor-6 (TRAF6) (Subhramanyam et al., 2019), which then activates the transforming growth factor-β-activated kinase-1 (TAK1) complex. This intricate signaling leads to the activation of two major pro-inflammatory pathways, the NF-κB and MAPK, resulting in the transcription of pro-inflammatory genes that contribute to AD pathogenesis (Kawai and Akira, 2007; Subhramanyam et al., 2019). Similarly, TLR2 also plays a critical role in the innate immune system, activating MyD88-mediated signaling (Cui et al., 2020). Interestingly, TLR2-mediated autophagy has been shown to regulate microglial M1/M2 phenotype switching, a process that could potentially modulate the inflammatory responses in AD (Ma et al., 2020). Autophagy can be stimulated by TLR2 signaling, suggesting a potential mechanism for controlling microglial phenotype and, thus, the neuroinflammatory response in AD. In summary, while the LPS-TLR4 complex instigates pro-inflammatory signaling pathways linked to AD, TLR2 signaling might modulate microglial phenotypes and AD’s neuroinflammatory pathogenesis through autophagy. These findings provide valuable insights into the mechanisms of microglial activation in AD and highlight potential therapeutic targets for this devastating neurodegenerative disease.
3.1.2. NF-κB signaling pathway
NF-κB is a pivotal transcription factor in the regulation of microglial activation, a process significantly involved in the inflammatory response seen in AD. The NF-κB p65/p50 subunits, in particular, play a major role in promoting M1 microglial activation. Conversely, inhibiting these subunits can suppress the transcription of inflammatory genes and promote a shift in microglia polarization toward the M2 phenotype (Zhang et al., 2013). This has been substantiated by the identification of numerous modulators that can induce a M1 to M2 phenotype shift in microglia by inhibiting NF-κB, further emphasizing the crucial role this pathway plays in microglial polarization and, by extension, in the inflammatory component of AD pathogenesis (Kawai and Akira, 2007). In the absence of stimulation, NF-κB remains inactive in the cytoplasm by interacting with inhibitors of NF-κB (IκB) proteins (Kawai and Akira, 2007). However, when stimulated by the TAK1 complex, the IκB kinase (IKK) complex catalyzes the phosphorylation of IκB proteins. Phosphorylated IκBs are then polyubiquitinated and degraded, which allows NF-κB p50/p65 to translocate into the nucleus (Kawai and Akira, 2007). There, they bind to target promoters and promote the transcription of pro-inflammatory genes, a process that has been implicated in the chronic neuroinflammation seen in AD (Mulero et al., 2019). In summary, NF-κB plays a crucial role in microglial polarization and the regulation of inflammatory gene transcription, both of which are significant in the context of AD. Greater understanding of the pathways influencing NF-κB activation and the mechanisms by which it governs microglial polarization may pave the way for the development of novel therapeutic strategies for AD and other neuroinflammatory diseases.
3.1.3. JAK/STAT signaling pathway
The JAK/STAT signaling pathway is integral in orchestrating immune and inflammatory responses, and has been implicated in the pathogenesis of neurodegenerative diseases, including AD (Ruganzu et al., 2021). Specifically, STAT1 and STAT3 have been identified as key players in mediating M1 microglial polarization by promoting the production of pro-inflammatory cytokines and chemokines (Lan et al., 2017). On the other hand, the activation of STAT6 by IL-4 is associated with the promotion of microglial polarization toward the M2 phenotype, a state considered to be neuroprotective due to its anti-inflammatory effects. This anti-inflammatory state is particularly significant as it could potentially mitigate the neuroinflammatory processes seen in AD (Lan et al., 2017). In terms of the mechanistic process, activation of the JAK/STAT pathway results in JAK-induced phosphorylation of STAT family proteins (Cianciulli et al., 2017). These phosphorylated STAT proteins then translocate to the nucleus and initiate the transcription of target genes, one of which includes the suppressor of cytokine signaling (SOCS) family (Xin et al., 2020). The SOCS family proteins serve as key negative feedback regulators, inhibiting the phosphorylation of JAK (Durham et al., 2019). Notably, numerous studies have shown that upregulated SOCS1 and SOCS3 could attenuate inflammatory responses and contribute to the mitigation of AD-associated neuroinflammation by promoting a shift in microglia from the M1 to M2 phenotype (Ruganzu et al., 2021). Therefore, these findings underscore the significance of the JAK/STAT pathway in the context of AD, by regulating microglial polarization and subsequent inflammatory responses. This intricate interplay between the JAK/STAT signaling pathway and microglial polarization offers potential therapeutic avenues for AD.
3.2. Anti-inflammatory (M2) polarization
3.2.1. AMPK signaling pathway
Inflammatory responses are known to trigger an increase in intracellular Calcium (Ca2+) influx, which initiates a series of biochemical reactions crucial in the context of microglial activation and AD pathogenesis. Specifically, the increased Ca2+ influx binds to calmodulin (CaM), thereby activating the highly conserved Ca2+/CaM kinase cascade, including Calcium/calmodulin-dependent protein kinase kinase-β (CaMKKβ) (Marcelo et al., 2016; Dalal et al., 2020). Once activated, CaMKKβ phosphorylates AMP-activated protein kinase (AMPK), a central energy sensor in many tissues including the brain (Liu and Chern, 2015; Marcelo et al., 2016). It’s worth noting that AMPK plays a pivotal role in microglial polarization, particularly in shifting microglia from the pro-inflammatory M1 phenotype to the neuroprotective M2 phenotype (Wang J. et al., 2018). This process is particularly relevant in the context of AD, where a shift toward M2 phenotype could potentially mitigate neuroinflammation and slow disease progression.
In addition to CaMKKβ, liver kinase B1 (LKB1), another upstream kinase, is involved in phosphorylating and activating AMPK. This activation is prompted by an increase in the AMP/ATP ratio, a common occurrence during cellular stress situations such as neuroinflammation seen in AD (Deng et al., 2019). Subsequently, AMPK activates peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) directly and also stimulates the production of NAD+, leading to the activation of Sirtuin 1 (Sirt1). Sirt1 then deacetylates and activates PGC-1α, a master regulator of mitochondrial biogenesis and function, whose dysregulation has been implicated in AD (Fernandez-Marcos and Auwerx, 2011). Originally, PGC-1α was identified through its interaction with peroxisome proliferator-activated receptor gamma (PPAR-γ), which consequently led to an increase in PPAR-γ transcriptional activity (Zhang et al., 2020). This process is significant, as PPAR-γ activation has been associated with beneficial effects in AD, including reduced neuroinflammation and Aβ burden.
3.2.2. PI3K/AKT signaling pathway
The Serine and Threonine kinase AKT, also known as Protein Kinase B (PKB), plays a critical role in the regulation of microglial polarization (Manning and Toker, 2017). The activation of AKT is stimulated by G-protein-coupled receptors (GPCRs) or receptor tyrosine kinases (RTKs), culminating in the activation of phosphoinositide 3-kinase (PI3K). PI3K subsequently phosphorylates the T308 and S473 residues of AKT, leading to its full activation (Manning and Toker, 2017). Interestingly, low levels of PI3K/AKT signaling are associated with the pro-inflammatory M1 phenotype, while enhanced PI3K/AKT signaling is linked to the anti-inflammatory M2 phenotype (Linton et al., 2019). The PI3K/AKT pathway also maintains extensive interactions with other major signaling pathways, thereby influencing diverse cellular functions. For instance, it operates downstream of the NF-κB pathway, promotes neuroinflammatory responses in AD (Yu et al., 2017). Additionally, the activation of PI3K/AKT pathway is facilitated by phosphorylated Janus Kinase (JAK), another key modulator of immune responses (Gao et al., 2018). AKT can indirectly inhibit AMPK activation by promoting ATP production or directly phosphorylate AMPK, which inhibits LKB1-mediated phosphorylation of AMPK (Manning and Toker, 2017). This interaction is particularly noteworthy considering AMPK’s role in microglial polarization and AD pathogenesis.
Furthermore, the PI3K/AKT pathway and the extracellular signal-regulated kinase (ERK) pathway exhibit mutual inhibition, adding another layer of complexity to the regulation of cellular functions, including cell proliferation, differentiation, metabolism, and immune responses (Manning and Toker, 2017). In summary, the PI3K/AKT pathway’s intricate crosstalk with other signaling pathways and its role in microglial polarization underscores its potential as a therapeutic target in neuroinflammatory diseases such as AD. Further exploration of these molecular interactions may yield novel insights into AD pathogenesis and treatment strategies.
3.2.3. PPAR-γ signaling pathway
Peroxisome proliferator-activated receptor-gamma (PPAR-γ), a ligand-inducible transcription factor belonging to the nuclear receptor superfamily, is significantly expressed in microglia and plays a pivotal role in AD pathogenesis (Carta and Pisanu, 2013). The regulatory function of PPAR-γ in microglia involves modulation of both pro- and anti-inflammatory cytokines and up-regulation of the M2 phenotype marker, Arginase-1 (Arg-1) (Ahmadian et al., 2013; Pisanu et al., 2014; Xu et al., 2015; Song et al., 2016; Song and Suk, 2017). Significantly, the activation of PPAR-γ signaling by Interleukin-4 (IL-4) drives microglial polarization toward the M2 phenotype (Jiang et al., 2020). Moreover, PPAR-γ serves as a crucial counter-regulatory mechanism, mitigating neuroinflammation by antagonizing the action of pro-inflammatory transcription factors such as AP-1, NF-κB, and STAT1 (Jacobi et al., 2012). For instance, PPAR-γ activation can disrupt the NF-κB signaling pathway by inhibiting the nuclear translocation of the p65 subunit or competing with NF-κB co-activators (Yang Z. et al., 2019). Interestingly, recent research has also shed light on the role of PPAR-γ in autophagy regulation in microglia. Studies indicate that PPAR-γ antagonism promotes the transition of microglia from M1 to M2 polarization by enhancing autophagy via the LKB1/AMPK signaling pathway (Ji et al., 2018). Given the involvement of impaired autophagy in the accumulation of Aβ and tau proteins, the role of PPAR-γ in autophagy regulation further underscores its potential as a therapeutic target in AD.
3.2.4. Notch signaling pathway
The Notch signaling pathway is a highly conserved, cell-to-cell communication mechanism with fundamental roles in various cellular processes, including proliferation, differentiation, stem cell maintenance, and apoptosis (Nowell and Radtke, 2017). The pathway comprises four Notch receptors (NOTCH 1–4) and five ligands from the Delta-Serrate-Lag (DSL) family, including Jagged 1 (JAG1), JAG2, and Delta-like 1, 3, and 4 (DLL1, DLL3, DLL4). These Notch receptors are transmembrane proteins, synthesized in the endoplasmic reticulum and transported to the plasma membrane. In response to ligand binding from an adjacent cell, the Notch receptor undergoes a proteolytic cleavage, releasing the Notch intracellular domain (NICD) (Majumder et al., 2021). This NICD translocates into the nucleus where it acts as a transcriptional co-activator, inducing the expression of target genes and regulating cellular processes. In the context of microglia, the Notch signaling pathway has been recognized for its role in dictating microglial function and polarization (Wu et al., 2018). In AD, an imbalance in microglial polarization, favoring the pro-inflammatory M1 state, contributes to chronic neuroinflammation, exacerbating neuronal damage and disease progression. Notch signaling, through its influence on microglial polarization, might be involved in this process, possibly serving as a potential therapeutic target for rebalancing microglial states and mitigating neuroinflammation in AD. Furthermore, studies have suggested that Notch signaling might directly contribute to AD pathology, with aberrant Notch activation observed in AD models and patient samples. This aberrant activation might influence the production and clearance of Aβ plaques.
3.3. The crucial role of P2 × 7 receptor in microglial M1/M2 phenotypic balance and AD progression
Microglia showcase a diverse range of activation states rather than being exclusively confined to the traditionally defined M1 and M2 phenotypes. This nuanced view is substantiated by recent transcriptomic analyses that illuminate the existence of several intermediate phenotypes bridging the classical M1 and M2 states (Ransohoff, 2016). Moreover, microglia have the ability to concurrently demonstrate both pro-inflammatory and phagocytic functions, indicating that these two functions are not mutually exclusive but may interact dynamically (Masuda et al., 2020). Within this context, the P2 × 7 receptor (P2 × 7R), a purinergic receptor abundantly expressed in microglia, plays a pivotal role. Intriguingly, P2 × 7R has been observed to bolster phagocytosis even in the absence of an external ATP agonist (Gu et al., 2010, 2011, 2012; Gu and Wiley, 2018). This unique function is seemingly regulated by the closed state of the receptor and involves residues 306–320 of P2 × 7R binding to extracellular debris, bacteria, and apoptotic cells (Gu et al., 2011). Moreover, it includes interactions of intracellular regions of P2 × 7R with non-muscle myosin heavy chain IIA (NMMHC IIA) and the cytoskeleton, which in turn facilitate the internalization of material into the cell (Gu et al., 2010, 2009). This phagocytic capacity can be hindered by cytochalasin D or a monoclonal antibody targeting the extracellular domain of P2 × 7R, emphasizing the receptor’s indispensable role in this process. Furthermore, the synthesis and surface expression of P2 × 7R were identified as crucial for the phagocytic function of human monocytic cells, and the interruption of these processes by siRNA led to a marked impairment of phagocytic function (Gu et al., 2011).
When extracellular ATP levels surge transiently, the P2 × 7 receptor transitions from acting as a scavenger receptor. This triggers an upswing in autophagy, indicated by a transient elevation in levels of the autophagosomal lipid marker LC3-II, resulting in the degradation of materials and pushing microglia toward a mixed M1/M2 activation state (Gu and Wiley, 2018; Campagno and Mitchell, 2021). The dynamic equilibrium between these M1 and M2 states is pivotal in microglia’s transformation into disease-associated microglia (DAM), a distinct state seen in neurodegenerative diseases like AD (Orre et al., 2014; Keren-Shaul et al., 2017). As DAMs recognize neurodegeneration-associated molecular patterns (NAMPs) such as Aβ plaques, they are believed to exhibit a hybrid M1/M2 phenotype (Deczkowska et al., 2018). Emerging evidence suggests that P2 × 7R’s modulation of the M1/M2 balance in microglia may impact the onset and progression of the DAM state, thereby potentially influencing the course of neurodegenerative diseases (Illes et al., 2020). Considering the complex roles P2 × 7R plays in molding microglial phenotypes and DAM states, P2 × 7R antagonists have displayed promise in ameliorating various neurodegenerative conditions (Beamer et al., 2017; Fabbrizio et al., 2017; Wang et al., 2017; Domercq and Matute, 2019; Illes et al., 2019a). Notably, selective P2 × 7R antagonists can block the receptor’s channel activity without disrupting its phagocytic function, offering a potential mechanism to restore a balance between inflammation and phagocytosis (Fletcher et al., 2019).
Under pathological conditions, persistent ATP stimulation activates P2 × 7R (Burnstock et al., 2011; Li et al., 2015; Illes et al., 2019b). Within an inflammatory milieu (M1 phenotype), this activation prompts a cascade of cellular responses, beginning with the interaction of Toll-Like Receptor 4 (TLR4) with lipopolysaccharide (LPS) and culminating in ATP-dependent stimulation of P2 × 7R. This interaction subsequently instigates NLRP3 inflammasome-mediated caspase-1 activation and secretion of the pro-inflammatory cytokine IL-1β (Perregaux and Gabel, 1998; Muñoz-Planillo et al., 2013). This process is primarily stimulated by a decrease in intracellular potassium levels, illustrating the intricate ion regulatory role of P2 × 7R (Di Virgilio et al., 2017, 2018; Madry et al., 2018). P2 × 7R activation can also lead to the release of other pro-inflammatory cytokines, such as IL-6 and TNF-α, further accentuating the inflammatory state (Shieh et al., 2014).
In summary, microglia present a complex activation spectrum that extends beyond the traditional M1 and M2 phenotypes, exhibiting multiple intermediate states (Figure 2). P2 × 7R significantly influences this phenotypic diversity by coordinating pro-inflammatory and phagocytic responses. Further, P2 × 7R is implicated in the progression of DAM states, notably in neurodegenerative conditions. Understanding the intricate role of P2 × 7R in managing microglial phenotypes and its therapeutic potential necessitates further research to develop effective strategies for combating AD.
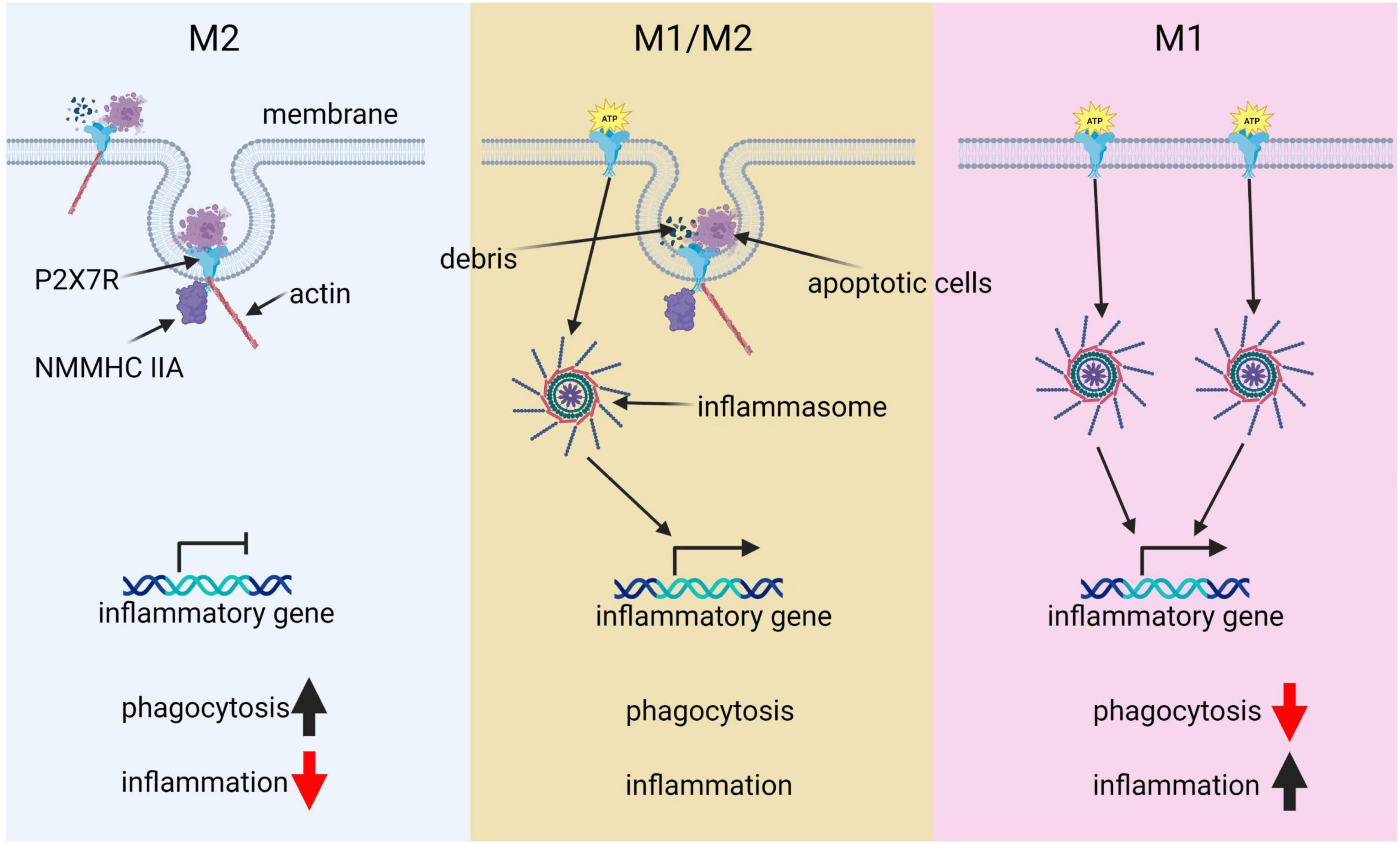
Figure 2. The pivotal role of P2 × 7R in microglial polarization. This diagram illustrates the key role of P2 × 7R in shaping microglial polarization states and its influence on immune responses. Under normal conditions, P2 × 7R interacts with extracellular debris such as apoptotic cells, as well as intracellular cytoskeleton components like actin and NMMHCIIA. This interaction induces phagocytic activity, resulting in minimal inflammation and predominance of the M2 state. However, transient ATP stimulation alters this balance: P2 × 7R not only drives phagocytosis but also gets activated by ATP, triggering downstream immunological inflammasome pathways and promoting inflammatory responses, thereby leading to a balance between M1 and M2 states. In a pathological condition, chronic ATP binding and activation of P2 × 7R shifts the cell primarily toward a pro-inflammatory and phagocytosis-suppressed phenotype, characteristic of the M1 state.
4. Phagocytosis of microglia in AD
Microglia play a pivotal role in the pathogenesis of AD by phagocytosing various targets such as apoptotic neurons, bacteria, lipoproteins, and Aβ (Hansen et al., 2018). The effect of microglia on AD progression depends on the specific substrate being phagocytosed. Microglial phagocytosis of Aβ is essential for clearing Aβ deposits and is considered a neuroprotective mechanism (Ries and Sastre, 2016). Microglia engulf Aβ through various receptors, such as Tyro3/Axl/Mer receptor tyrosine kinases (TAM) (Huang et al., 2021), scavenger receptor A (SRA), CD36 (Frenkel et al., 2013; Lucin et al., 2013), transporting it to lysosomes, where it is compressed into dense core plaques and toxic substances. Overactivation or phagocytic dysfunction of microglia can lead to Aβ accumulation and promote AD progression. Excessive phagocytosis of normal neuronal synapses by microglia is a critical factor contributing to cognitive decline in AD. Aβ binding to complement component C1q can activate the classical complement pathway, resulting in excessive microglial phagocytosis of synapses. This complement-dependent pathway and excessive microglial activation, leading to synaptic loss, are associated with cognitive dysfunction in AD (Schafer et al., 2012). Inhibiting C1q, C3, and C3R expression can effectively reduce the number of activated microglia and early synaptic loss (Hong et al., 2016). Furthermore, microglial phagocytosis of the extracellular matrix (ECM) surrounding synapses promotes synaptic structural remodeling and is involved in memory consolidation. IL-33 mediates experience-dependent neuron-microglia communication, promoting hippocampal dendritic spine formation, synaptic remodeling, and ECM phagocytosis, all essential for memory consolidation. Loss of IL-33 results in impaired microglial phagocytosis of ECM, accompanied by the accumulation of ECM proteins related to synapses (Végh et al., 2014b; Nguyen et al., 2020). Clearing ECM and administering exogenous recombinant IL-33 have been shown to improve AD model mice (Végh et al., 2014a; Fu A. K. Y. et al., 2016). In this review, we will provide a comprehensive analysis of the genes involved in microglial phagocytic function, their implications for AD pathogenesis, and potential therapeutic targets.
4.1. Phagocytosis of apoptotic neurons by microglia
In AD, microglia rapidly engulf dying or apoptotic neurons, suppressing further inflammation (Lazdon et al., 2020). The phagocytic process of microglia can be divided into three stages: “Find me,” “Eat me,” and “Digest me.” “Find me” signals are cell factors released by apoptotic cells that attract phagocytic cells. These signals include CX3CR1, LPC, S1P, and nucleotides, which bind to corresponding receptors on phagocytic cells (CX3CR1, G2A, S1P-R1/5, and P2Y2, respectively), promoting their migration toward apoptotic cells. “Eat me” signals are recognized by phagocytes following “Find me” signals. During phagocytosis, apoptotic cells expose “Eat me” signals, enabling phagocytes to distinguish them from healthy cells. Phosphatidylserine (PtdSer) is a crucial “Eat me” signal, which can directly or indirectly bind to phagocyte surface receptors and mediate downstream signaling pathways (Arandjelovic and Ravichandran, 2015; Figure 3). These include: (1) direct binding of PtdSer to PtdSer receptors such as BAI1, which recognizes PtdSer and binds to intracellular ELMO1 and Dock, activating downstream Rac1, causing actin and other cytoskeleton rearrangements, and promoting phagocytosis of apoptotic cells (Arandjelovic and Ravichandran, 2015); (2) PtdSer activates integrin receptors through bridging molecules such as MFG-E8, and activates downstream Rac1 via the CrkII-Dock180-ELMO signaling pathway (Arandjelovic and Ravichandran, 2015); (3) PtdSer activates TAM receptors through bridging molecules such as Gas6 or Protein S, and downstream signaling pathways may induce cytoskeleton remodeling by activating PI3K or PLC (DuBois et al., 2020; Lew et al., 2020; Rosin et al., 2021); and (4) in addition to PtdSer, calcium-binding protein (calreticulin) or complement C1qa can also activate LRP1/CD91 to bind to scaffold protein GULP1 and activate downstream Rac1 (Cockram et al., 2019; Hammond et al., 2020). “Digest me” signals mediate the processing of phagocytosed apoptotic cells, which requires the maturation of phagosomes (Han et al., 2021).
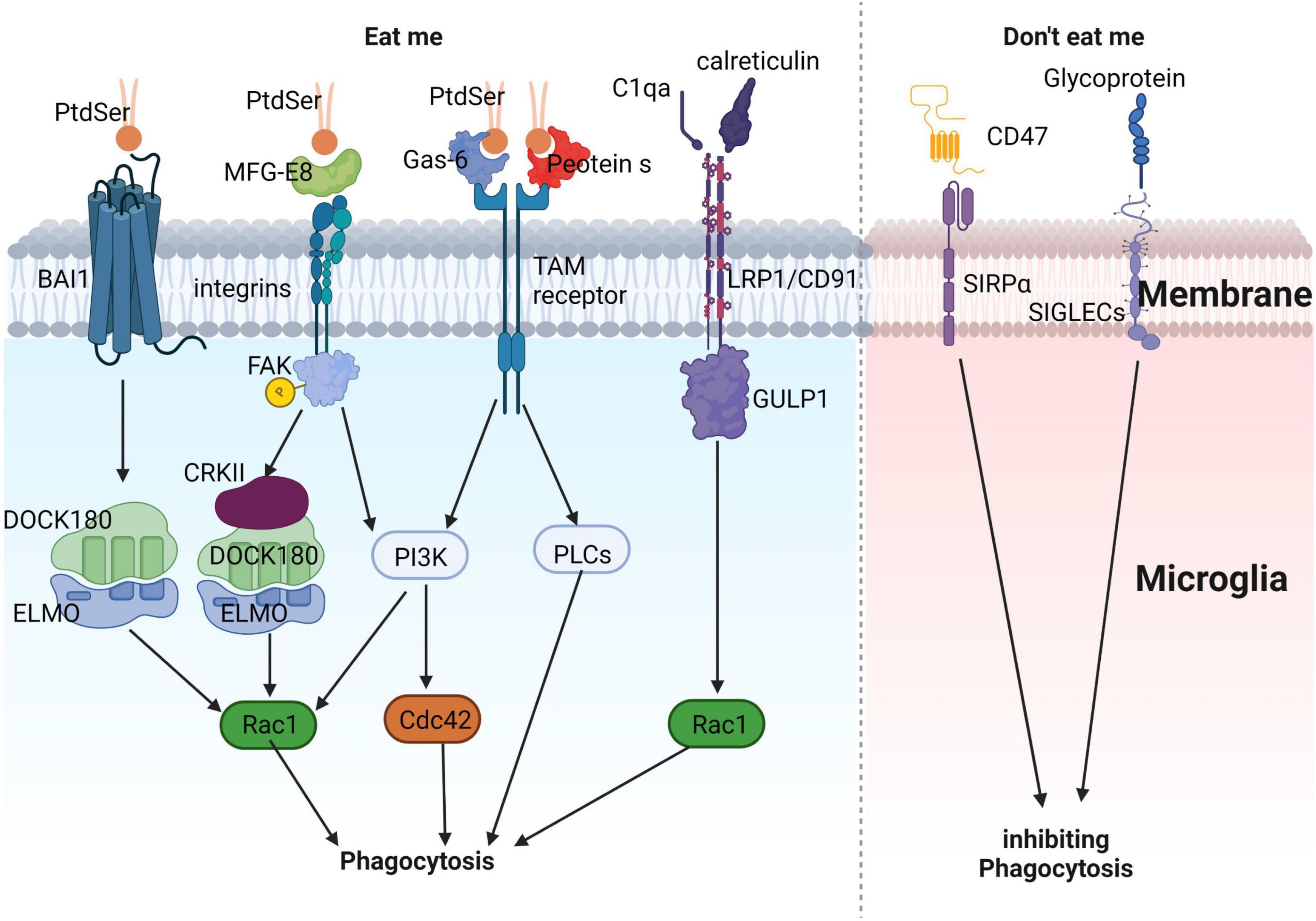
Figure 3. Illustrates the process of microglial phagocytosis of apoptotic neurons. The detection of dying neurons by microglia is mediated by the release of “eat-me” signals from apoptotic neurons. PtdSer interacts either directly with the microglial BAI1 receptor or indirectly through MFG-E8, Gas-6, and protein S. These proteins can engage microglial receptors such as integrins and TAM receptors, ultimately promoting the phagocytosis of apoptotic neurons. Apart from PtdSer, other eat-me signals include calreticulin and complement component C1q. These molecules can associate with LRP1/CD91 to facilitate phagocytosis. In contrast, “don’t-eat-me” signals, which inhibit phagocytosis, consist of sialylated glycoproteins and lipids, as well as the CD47 protein. These components can interact with microglial receptors like SIGLECs and SIRPα, respectively, to suppress phagocytosis, ensuring a fine-tuned regulation of this crucial process.
”Don’t eat me” signals inhibit phagocytic cells from exerting their phagocytic function. CD47, a transmembrane protein expressed in most mammalian cells, including neurons, inhibits phagocytic activity via the signal-regulatory protein alpha (SIRPα) on phagocyte surfaces. CD47 also inhibits microglial-mediated synapse clearance during development (Butler et al., 2021). Sialic acid proteins inhibit neuronal phagocytosis by activating sialic acid-binding immunoglobulin-like lectins (SIGLECs) on microglial surfaces (Brown and Neher, 2014).
Additionally, a novel phagocytic pathway called LC3-associated phagocytosis (LAP) has recently been reported. LAP plays a role in engulfing both apoptotic and live cells. Injecting apoptotic cells into LAP-deficient animals promotes lupus-like disease, increases pro-inflammatory cytokine production, and decreases anti-inflammatory cytokine production (Martinez et al., 2016). LAP inhibits the inflammatory response during engulfment of apoptotic cells in vitro and in vivo. During LAP, the engulfment body recruits components of the autophagic machinery, catalyzes the lipidation of LC3 on the engulfment body membrane into phosphatidylethanolamine, and induces engulfment body maturation and cargo degradation. LAP is induced by binding of TLR1/2, TLR2/6, or TLR4, Fc receptors, and apoptotic cell receptor TIM4 on the engulfment body (Heckmann et al., 2017). Unlike canonical autophagy, LAP does not require components of the preinitiation complex (ULK1, FIP200, ATG13) to initiate autophagy in response to nutrient stress, but it requires a unique Beclin-1 (BECN1) and VPS34 initiation complex that lacks ATG14 but includes Rubicon (Heckmann et al., 2017). Phagocytosis and clearance of substrates are important pathways for microglia to maintain tissue homeostasis.
4.1.1. C1q: facilitator of microglial phagocytosis and synaptic pruning in AD
C1q is a critical protein that regulates the production of inflammatory cytokines and aids in the removal of apoptotic neurons and neuronal blebs by microglia. Direct binding to Aβ and being the initial constituent of the classical complement pathway are among the functions of C1q (Jiang et al., 1994). However, complement activation can lead to neuronal lysis, resulting in inflammation, neuronal damage, and impaired neuronal integrity (Tenner, 2001). C1q’s involvement in the initiation of the classical complement pathway results in several downstream effects, including opsonin C3b deposition on target cell surfaces, pathogen lysis, and phagocytic cell recruitment (Fraser and Tenner, 2008). During the early stages of cell death, C1q’s role in the central nervous system is protective, increasing microglial clearance of apoptotic cells and suppressing proinflammatory cytokines (Fraser et al., 2010). However, in AD pathology, C1q’s role is more complex, with associations with both fibrillar Aβ plaques and tangles (Afagh et al., 1996; Shen et al., 2001). C1q positive Aβ plaques are associated with reactive astrocytes or microglia, which are frequently associated with the neurodegenerative process (Dickson, 1997; Griffin et al., 1998). Increased recruitment of activated glial cells may cause enhanced inflammation and contribute to the relationship between C1q and AD pathology (Fonseca et al., 2004a). Furthermore, C1q enhances phagocytosis and binding to apoptotic cells, which is a complement and microglia-dependent pathway that removes excess synapses, overactivation of this pathway leads to synaptic loss in AD (Korb and Ahearn, 1997; Webster et al., 2000; Hong et al., 2016). C1q inhibition decreases the number of phagocytic microglia and the amount of initial synapse loss by inhibiting C1q, C3, or the microglial complement receptor CR3 (Fonseca et al., 2004b; Hong et al., 2016; Shi et al., 2017). C1q is believed to exert toxic effects on hippocampal long-term potentiation (LTP) and synapses affected by soluble Aβ (Hong et al., 2016). In summary, C1q’s role in AD pathology is complex, with both protective and harmful effects.
4.1.2. TLR-4: essential modulators of microglial phagocytosis and axon clearance in the CNS
Studies have shown that the phagocytosis of myelin by microglia and the clearance of apoptotic cells contribute significantly to the inhibition of inflammation and restoration of the CNS (Fadok et al., 1998; Franklin and Kotter, 2008). TLRs also play a critical role in the clearance of oligomeric, monomeric, and fibrillar Aβ by microglia (Reed-Geaghan et al., 2009). Additionally, TLRs such as TREM2, PtdSer receptor T cell immunoglobulin mucin-4, and soluble linking proteins MFG-E8 and Gas6 are crucial for microglial phagocytosis of apoptotic cells (Miyanishi et al., 2007; Grommes et al., 2008; Linnartz and Neumann, 2013). Genetic and pharmacological interruption of TLR4 blocks the induction of the type-1 interferon response, restricting the phagocytosis of ex-vitro axon remains, while TLR4-dependent microglial clearance of unmyelinated axon remains facilitates the outgrowth of axons (Takahashi et al., 2005; Rajbhandari et al., 2014). TLR4-mediated signaling may also be affected by purinergic receptors such as the P2 × 7 receptor (P2 × 7R), which may lead to the clearance of microglia-degenerated axons (Takahashi et al., 2005; Rajbhandari et al., 2014). Elimination of TLR4 in adult mice results in impaired microglial phagocytosis of CNS axons, particularly neurons undergoing Wallerian degeneration in a model of dorsal root axotomy (Rajbhandari et al., 2014). These findings suggest that TLRs and purinergic receptors play an essential role in microglial phagocytosis and axon clearance, which may contribute to the restoration of the CNS.
4.1.3. ANXA1: regulator of microglial phagocytosis and blood-brain barrier integrity
ANXA1 is a mediator of glucocorticoid anti-inflammatory effects in the peripheral system, contributing to the efficient elimination of apoptotic neuron-form cells (Perretti and D’Acquisto, 2009; McArthur et al., 2010). It also plays a crucial role in maintaining the tightness of the blood-brain barrier (BBB) by being present in the BBB endothelium and microglial cells (Solito et al., 2008; Cristante et al., 2013). Microglia can synthesize and release ANXA1, which is associated with regulating leukocyte extra-vasation, phagocytosis, and glucocorticoid actions (Young et al., 1999; Perretti et al., 2002; Yona et al., 2004; Buckingham et al., 2006). ANXA1 can be upregulated in human microglia surrounding Aβ plaques and has been identified as a receptor for the formyl peptide receptor-like 1 (FPRL1), which also interacts with Aβ (Young et al., 1999; Buckingham et al., 2006). The attachment of ANXA1 to FPRL1 regulates microglial phagocytosis and proinflammatory cytokine release, enhancing the phagocytic action of microglia (Young et al., 1999; Le et al., 2001; Pan et al., 2011). Therefore, ANXA1 is a critical factor in maintaining the integrity of the BBB and regulating microglial function, which may have implications for neuroinflammatory conditions.
4.2. Trogocytosis of microglia
Trogocytosis is a process by which microglia nibble off portions of other live cells, primarily synapses, unlike phagocytosis, which targets dead cells (Weinhard et al., 2018). Microglia use trogocytosis to reshape neuronal synapses, mediating synaptic remodeling (Lim and Ruthazer, 2021). While the molecular mechanisms of trogocytosis have been well-studied in peripheral immune cells, recent research has shed light on the role of trogocytosis in microglia. The intercellular receptor-ligand interaction is crucial for trogocytosis, and actin and PI3K are involved (Bettadapur et al., 2020). In the immune system, trogocytosis is associated with the downregulation of the “Don’t eat me” signal CD47-SIRPα and the upregulation of the CD11b/CD18 integrin pathway (Matlung et al., 2018). Receptors play a critical role in determining the activation of immune cells in phagocytosis or trogocytosis. Chimeric antigen receptor-phagocytic receptors (CAR-Ps) can guide macrophages to ingest target cells through trogocytosis rather than phagocytosis, and this process depends on the ITAM-containing intracellular signaling domain (Morrissey et al., 2018). The specificity of receptor interaction plays an important role in mediating phagocytosis or trogocytosis. Conflicting results exist regarding the role of the complement pathway in microglial trogocytosis. The absence of CR3 did not significantly affect trogocytosis in microglia in vitro brain slice cultures of CR3 knockout mice, suggesting that complement pathway participation is not required for microglial trogocytosis (Weinhard et al., 2018). However, in vivo studies in African clawed frogs indicate that trogocytosis in microglia required the complement pathway, and C3 mediate microglial trogocytosis (Hodge et al., 2019; Lim and Ruthazer, 2021). Further research on the molecular mechanisms of trogocytosis in different cell types and pathological states is necessary to better understand this unique process and develop potential therapeutic strategies.
4.3. Comparation between phagocytosis and trogocytosis
In organisms, phagocytosis and trogocytosis are two primary ways that macrophages clear cells or particles. Phagocytosis is used for larger particles, such as dead cells, and involves engulfing the entire target. Trogocytosis, on the other hand, is used for smaller particles, generally less than 1 μm in size, and involves nibbling away at the target (Figure 4). If the target is too large to be internalized through phagocytosis, macrophages will surround the target, which is known as “phagocytic failure” (Chen et al., 2019). It’s important to note that macrophages involved in phagocytic failure do not digest the target through nibbling, so trogocytosis is not the result of phagocytic failure. The molecular mechanisms underlying phagocytosis and trogocytosis are also different. During phagocytosis, highly conserved pathogen-associated molecular patterns (PAMP) bind to FcγR and enter the cell, activating downstream signaling pathways that induce actin polymerization and formation of phagosomes. When FcγR binds to artificially designed antibodies, it can induce macrophages that originally have phagocytic activity to take up target cells through trogocytosis and induce non-professional phagocytic cells to exhibit trogocytic function (Morrissey et al., 2018). In the CNS, microglia are responsible for phagocytosing neuronal synapses (Schafer et al., 2012). The complement pathway is activated during this process, with C1q binding to and opsonizing apoptotic cells, pathogens, or debris. This triggers a cascade of proteases that result in the deposition of downstream complement protein C3. C3 deposited on dendrites and synapses can directly activate C3 receptors on microglia, leading to dendrite and synapse phagocytosis. The key step in complement pathway activation is the cleavage of C3 into C3a and C3b, where C3a recruits and activates microglia, and C3b opsonizes neurons and synapses (Butler et al., 2021). Studies have shown that excessive phagocytosis of weak synapses by complement-mediated microglia may lead to cognitive impairment during memory extinction (Butler et al., 2021). Conversely, knocking out C1q, C3, or CR3 in mice reduces synaptic pruning during development, leading to the production of excess synapses (Hong et al., 2016). Research on the molecular mechanisms underlying microglial trogocytosis in the CNS is still incomplete. It is unclear whether the complement pathway is involved in microglial trogocytosis. Further investigation into the molecular mechanisms of trogocytosis is of great significance for understanding the pathogenesis of neurodegenerative diseases and may provide new molecular targets for their treatment.
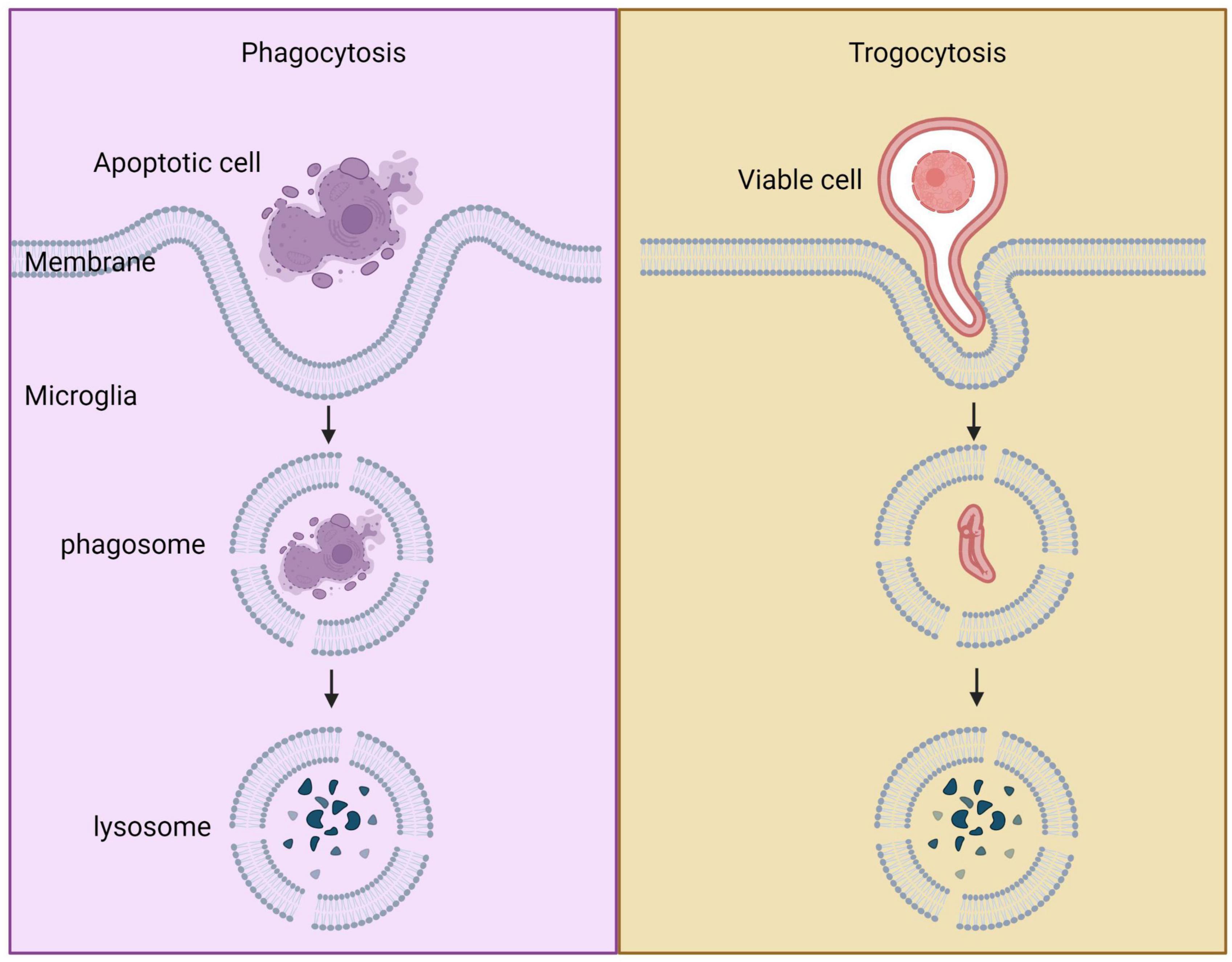
Figure 4. Comparison between phagocytosis and trogocytosis. Trogocytosis is a process where live cell targets are nibbled, whereas phagocytosis is a process where dead cell targets are engulfed.
4.4. Phagocytosis of Aβ
Current research has focused on understanding how Aβ is removed from the body. Aβ can enter peripheral circulation through various routes, such as transfer across the blood-brain barrier, the glymphatic system, or perivascular drainage (Shibata et al., 2000; Weller et al., 2008; Iliff et al., 2012; Iliff and Nedergaard, 2013). Microglia cells are known to play a role in phagocytosis of Aβ, including soluble oligomeric Aβ. Other cells, such as, astrocytes, olfactory ensheathing cells (OECs), and neurons, also contribute to degrading and clearing Aβ (Scheuner et al., 1996; Malm et al., 2010). Certain molecules, such as MFG-E8 and the TAM receptor family, aid in the phagocytosis process by binding to Aβ. However, the way in which Aβ aggregates can vary depending on the binding proteins and their interaction with Aβ.
4.4.1. MFG-E8: promotor of microglial phagocytosis of Aβ
MFG-E8 is a vital molecule that plays a crucial role in the process of phagocytosis, which involves the engulfment and elimination of apoptotic cells. This molecule binds to integrin molecules such as integrin β3 on the surface of phagocytes. Integrin receptors consist of paired α and β sub-units (Hynes, 2002; Barczyk et al., 2010) and are essential for various cellular functions such as adhesion, migration, growth, differentiation, and reprogramming in macrophages (Chen et al., 2009; Barczyk et al., 2010). Produced by macrophages, MFG-E8 helps to alleviate inflammation and promote efferocytosis, which is the process of clearing apoptotic cells (Kojima et al., 2017). Interestingly, there appears to be a direct link between MFG-E8 and transglutaminase 2 (TG2), which is also involved in cholesterol transfer and enhances phagocytosis (Hanayama et al., 2002; Boisvert et al., 2006). TG2 acts as a co-receptor of β3 integrin and attaches to extracellular MFG-E8 (Tóth et al., 2009). Recent studies have shown that microglia and astrocytes release MFG-E8 in response to stimuli from neurons triggered by Aβ (Neher et al., 2011; Kawabe et al., 2018). TG2 also contributes to phagocytosis, with research indicating that TG2 peritoneal macrophages play an essential role in recognizing apoptotic cells through MFG-E8 and integrin β3 (Akimov et al., 2000; Tóth et al., 2009). Furthermore, the TG2 protein mediates the attachment between MFG-E8 and the vitronectin receptor (VR) independently of TG enzyme activity (Tóth et al., 2009; Kawabe et al., 2018). Studies have shown that MFG-E8 reduces cell death in response to Aβ-induced neurons and enhances microglial phagocytosis of Aβ. MFG-E8 related therapy also activates the nuclear factor erythroid 2-associated factor 2 (Nrf2)-HO-1 pathway, which protects Aβ-activated neurons from cell death (Li et al., 2012). In summary, MFG-E8 is a critical molecule involved in phagocytosis and is produced by macrophages in response to inflammation. Its interaction with integrin molecules and TG2 contributes to the recognition of apoptotic cells and promotes their clearance. In the brain, MFG-E8 plays a crucial role in reducing neuronal cell death and enhancing microglial phagocytosis of Aβ, suggesting that it could be a potential therapeutic target for AD.
4.4.2. CD36: crucial for fibrillar Aβ phagocytosis but non-essential for soluble Aβ clearance
CD36, a member of the scavenger receptor class B family, is expressed on the surface of various cells such as neurons, macrophages, astrocytes, and monocytes (Yu and Ye, 2015). It plays an important role in the phagocytosis of fibrillar Aβ42 by binding to it (Wilkinson and El Khoury, 2012; Yu and Ye, 2015). Studies have shown that CD36 deficiency in mice prevents the accumulation of microglia in response to the injection of fibrillar Aβ (El Khoury et al., 2003), indicating that intracerebral microinjection of fibrillar Aβ can be used to study the molecular and cellular mechanisms of Aβ-mediated brain responses and screen for new therapies in CD36 knockout mice (El Khoury et al., 2003). In vitro experiments have also shown that antagonists of CD36 can efficiently block the phagocytosis of fibrillar Aβ42 by microglia (Koenigsknecht and Landreth, 2004). The expression of CD36 is significantly higher in monocyte-extracted macrophages activated by glatiramer acetate, which plays an important role in the clearance of Aβ (Koronyo et al., 2015). While CD36 can bind to soluble Aβ42, its role in the clearance of soluble Aβ is not essential (Frenkel et al., 2013; Sheedy et al., 2013). Knockdown or suppression of CD36 does not affect the continuous capability of microglia to phagocytose soluble Aβ42. The prolonged expression of additional scavenger receptors compensates for the loss of CD36 function (Wilkinson et al., 2011). However, the deficiency of class A1 scavenger receptors impairs the clearing of soluble Aβ via mononuclear phagocytes and accelerates the development of AD (Frenkel et al., 2013). Interestingly, the selective elimination of CD36 does not affect the uptake of soluble Aβ, suggesting that CD36 is not necessary for clearing soluble Aβ. However, CD36 is involved in activating mononuclear phagocytes through soluble Aβ, which may lead to the production of cytokines and reactive oxygen species (El Khoury et al., 2003; Stewart et al., 2010; Frenkel et al., 2013). This is similar to the role CD36 plays in the interaction between oxidized low-density lipoproteins and macrophages (Maxeiner et al., 1998).
4.4.3. LRP1: facilitators of Aβ clearance in AD pathology
Recent studies suggest that LDL-receptor-associated protein 1 (LRP1) plays a crucial role in discriminating between apoptotic and normal cells. Up-regulation of calreticulin (Calr) on the apoptotic cell surface enhances their interaction with LRP1 and promotes the activation of phagocytic cells, leading to successful engulfment of apoptotic cells (Gardai et al., 2005). LRP1 forms a complex with Calr, functioning as a collectin receptor on macrophages. The role of LRP1 has also been demonstrated in the clearance of Aβ and transmission of signals involved in the pathology of AD (Kanekiyo and Bu, 2014). However, expression of APOE4 can lead to less efficient microglial clearance of Aβ, thereby affecting Aβ metabolism and microglial response (Zhao et al., 2014; Krasemann et al., 2017). APOE isoforms compete for identical clearance paths into the brain and can affect Aβ metabolism, as evidenced by studies in APOE-deficient mice exhibiting reduced fibrillar plaque deposition and altered regional distribution of plaque pathology within the hippocampus (Verghese et al., 2013; Ulrich et al., 2018). In a mouse model, APOE isoforms modulate the metabolism of soluble amyloid beta protein (sAβ) within astrocytes and the interstitial fluid by competing for the LRP1-dependent cellular absorption pathway in astrocytes (Verghese et al., 2013). Additionally, APOJ modifies the capability of forming fibrils and alters Aβ-induced neurotoxicity. Both APOE and APOJ are generated in astrocytes, where they facilitate the transport and clearance of Aβ through the blood-brain barrier via the megalin/LRP-2 receptor. Exposure of astrocytes to Aβ fused with APOJ, α1-antichymotrypsin (ACT), APOE, and integration of serum amyloid protein (SAP) and supplement C1q leads to a significant decrease in astrocyte expression, while no microglial Aβ absorption is observed. These findings highlight the crucial role of LRP1 and APOE isoforms in the clearance of Aβ and the development of AD pathology (Mulder et al., 2014).
4.4.4. ABCA7: enhancer of Aβ clearance in AD pathology
ABCA7 is a cytosolic protein that plays a crucial role in phagocytosis, and its upregulation has been associated with increased phagocytosis in certain disorders (Jehle et al., 2006; Yancey et al., 2010). Recent genome-wide studies have revealed a significant relationship between ABCA7 and AD (Hollingworth et al., 2011). This transporter is predominantly expressed in microglia, where it protects the brain by regulating the secretion and deposition of Aβ (Kaminski et al., 2000; Koldamova et al., 2003; Wahrle et al., 2005). Studies have demonstrated that ABCA7–/– mice exhibit reduced phagocytic clearance of Aβ oligomers in the hippocampus. Moreover, upregulation of ABCA7 in the hippocampus of the AD brain and amyloidogenic mouse brain appears to contribute to the pathogenesis of Aβ (Fu Y. et al., 2016).
4.4.5. TNF-α: influencing microglial phagocytosis and Aβ production in AD
The link between neuroinflammation and AD pathogenesis is well-established through clinical and experimental evidence (Heneka et al., 2015; Heppner et al., 2015). Inflammatory molecules such as IL-6, IL-1β, and TNF-α have been implicated in AD pathology (Wyss-Coray and Rogers, 2012). TNF-α is known to play a critical role in AD pathogenesis as it is expressed in neurodegenerative diseases. Studies in 5 × FAD/TNF-α–/– mice show that the absence of TNF-α reduces Aβ in the brain. TNF-α influences Aβ production by decreasing the levels of β- and α-secretases in 5 × FAD/TNF-α–/– brains, and it also affects PS1-mediated activities by decreasing the levels of PS1-carboxyterminal particles. TNF-α defects lead to a decline in microglial and astrocyte activation, resulting in decreased phagocytic activities of macrophages, including responsiveness toward Aβ. Genetic deletion of TNF-α in 5 × FAD mice reduces amyloid plaque formation by reducing the production of Aβ through activation of PS1 and β-secretase, rather than enhancing Aβ clearance through phagocytosis. Paouri et al. showed that amyloid pathology was modulated by TNF-α using cells and glial from AD mice’s brain (Paouri et al., 2017a,b).
4.4.6. γ-secretase: dual role in Aβ production and impaired phagocytosis in AD
γ-secretase is an enzyme that plays a critical role in the development of AD. Alterations in gamma-secretase activity can affect the expression of scavenger receptors, including SR-A, CD36, and CD40, which are involved in the clearance of Aβ by microglia (Husemann et al., 2002; El Khoury et al., 2003; Yang et al., 2011). While microglia and macrophages share similar phagocytic capabilities, blood-derived microglia have been shown to exhibit greater phagocytic activity toward Aβ deposits (Simard et al., 2006). Presenilin 1 and 2 (PS1 and PS2) are important determinants of gamma-secretase catalytic localization and play a crucial role in APP processing to generate neurotoxic Aβ isoforms (Wolfe, 2008). Depletion of the PS1 and PS2 catalytic site leads to impaired soluble Aβ phagocytosis and clearance of insoluble Aβ plaques. PS2 deficiency has been shown to impair Aβ phagocytosis in peritoneal macrophages in vivo. Furthermore, γ-secretase has a dual role in AD, as cleavage of APP by γ-secretase is associated with the pathogenesis of AD and Aβ accumulation, while microglial activation in AD is mediated by γ-secretase. Mutations in PS alter γ-secretase activity, leading to microglial dysfunction and increased amyloid aggregation in AD (Farfara et al., 2011). Therefore, dysfunction in microglial clearance of Aβ may be linked to alterations in γ-secretase activity, particularly in the context of PS mutations that accelerate AD pathology.
4.4.7. RAGE: facilitator of Aβ-induced neurodegeneration and modulator of phagocytosis in AD
The receptor for advanced glycation end products (RAGE) is involved in the process of phagocytosis by acting as a receptor for PtdSer and may play a role in inflammation resolution (Friggeri et al., 2011; He et al., 2011). Additionally, RAGE can exist in a soluble form (sRAGE) that acts as a RAGE ligand (Santilli et al., 2009). RAGE can bind to the globular heads of C1q and enhance the phagocytosis of apoptotic cells mediated by C1q by forming a receptor complex with complement receptor 3 (CR3; CD11b/CD18) (Ma et al., 2012). When Aβ fibrils bind to RAGE, signal transduction is initiated through adaptor proteins on the cytoplasmic domain of RAGE (Yan et al., 1996). Oligomeric Aβ strongly activates RAGE in neurons and microglia, which leads to neurodegeneration (Yan et al., 2009; Origlia et al., 2010; Srikanth et al., 2011). Moreover, it has been suggested that scavenger receptor and macrophage receptor with collagenous structure (SR-MARCO) and RAGE form a network with FPRL1/FPR2 to initiate microglial signaling in response to Aβ (Slowik et al., 2012).
4.5. Phagocytosis of tau
Abnormal accumulation of hyper-phosphorylated tau protein contributes to neuronal death in AD (Medina and Avila, 2014). The CX3CL1/CX3CR1 signaling pathway is crucial in deactivating microglia and inhibiting the release of proinflammatory cytokines (Cardona et al., 2006; Biber et al., 2007; Ginhoux et al., 2010). Defects in this pathway result in the enhanced generation of proinflammatory molecules, which leads to the deterioration of Tau via microglia (Bolós et al., 2017). In vitro and in vivo studies have demonstrated that CX3CR1/CX3CL1 axis plays a significant role in phagocytosing Tau in microglia and is a contributing factor in AD development. Targeting CX3CR1 could be a potential strategy for clearing extracellular Tau (Bolós et al., 2017).
5. Inflammation
5.1. Neuroinflammation mediated by microglia in CNS
Neuroinflammation is the response of the CNS to endogenous and/or exogenous factors that disrupt normal cellular homeostasis (Maccioni et al., 2018). Microglia are essential for detecting various endogenous and exogenous signals in the CNS. Inflammatory signals are a category of stimuli that are classified as either danger-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), or molecular processes that alter homeostasis (Schafer et al., 2012). Microglia are also involved in the entire process from innate immunity to subsequent inflammation in the brain (Maccioni et al., 2018; Pang et al., 2022; Van Zeller et al., 2022). Pattern recognition receptors (PRRs), such as TLRs, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and AIM2-like receptors, are involved in the detection of DAMPs and PAMPs by microglia in the CNS (Freeman et al., 2017; Ma et al., 2021). Following detection of DAMPs or PAMPs, microglia become activated and release a cascade of cytokines and chemokines in response to the injury or pathological event, which serves to elicit an immune response and modulate the microenvironment (Wang Y.-J. et al., 2021).
There are three distinct phenotypes of microglia (Figure 5; Clayton et al., 2017). Under normal and non-aging circumstances, microglia exhibit a homeostatic phenotype within the adult brain. However, with advancing age, the expression of homeostasis markers within microglia gradually decreases, resulting in diminished functionality, including reduced proliferation, phagocytosis, branching, and cytokine secretion. This, in turn, can lead to the emergence of microglia with a dystrophic phenotype (von Bernhardi et al., 2015; Clayton et al., 2017). In neurodegenerative conditions, a specific subset of microglia known as DAM are observed to be present, which display altered gene expression profiles that are associated with increased inflammatory responses and reduced expression of genes involved in maintaining microglia homeostasis (Keren-Shaul et al., 2017; Krasemann et al., 2017).
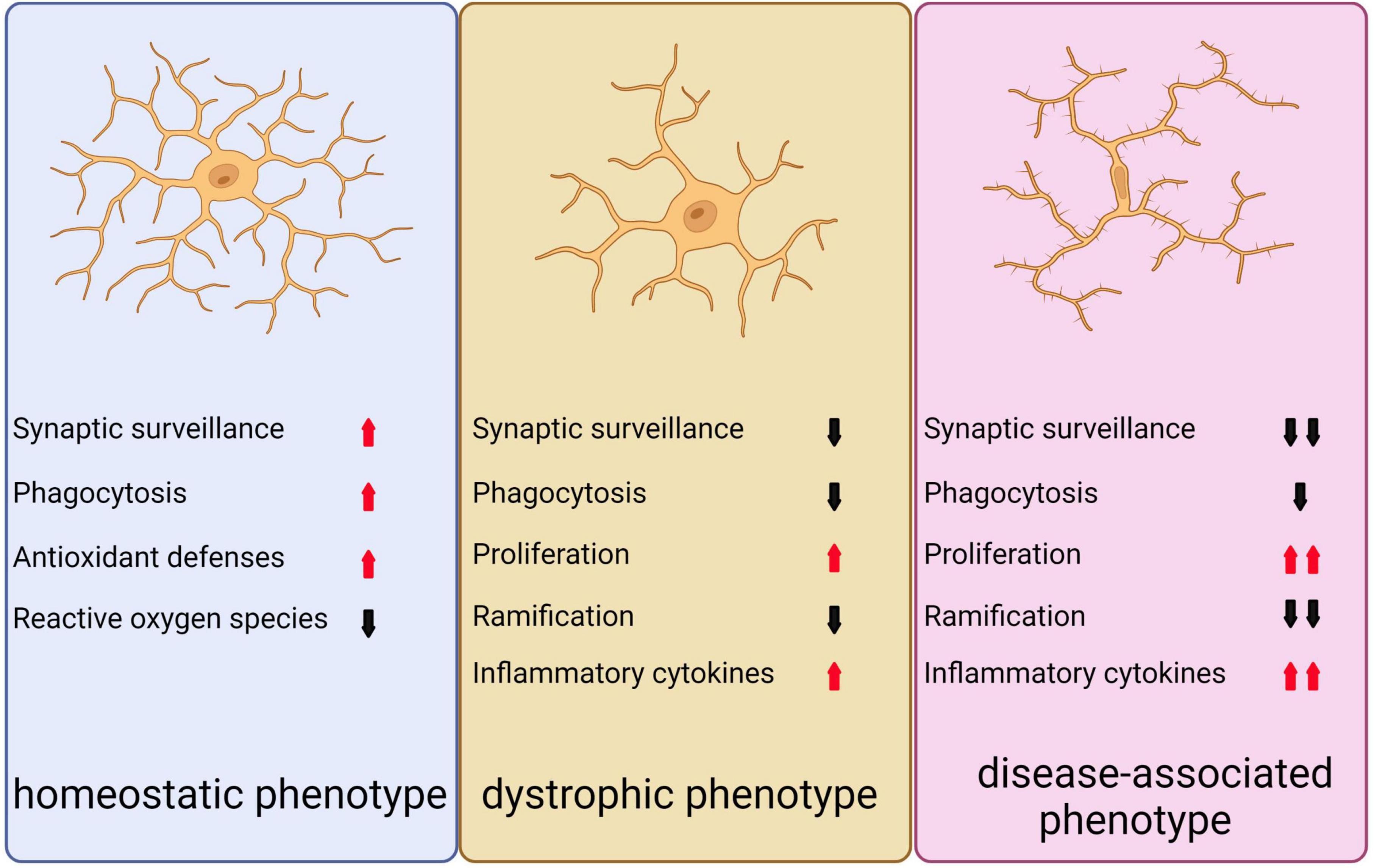
Figure 5. Depicts the three distinct microglial phenotypes. The first, known as the homeostatic phenotype, is generally observed in the adult brain under non-pathological, non-infected, and non-aged conditions. The second phenotype, referred to as the dystrophic phenotype, emerges during normal aging as homeostatic markers gradually diminish, leading to a decline in various microglial functions such as proliferation, phagocytosis, ramification, and cytokine secretion. The third phenotype, known as the disease-associated phenotype, is linked to neurodegeneration and presents an exacerbated form of the dystrophic phenotype, reflecting the diverse roles microglia can assume in different contexts.
Although the M1/M2 classification of microglia has recently been controversial, it may help explain the pathological relationship between inflammation and degenerative CNS diseases (Song and Suk, 2017; Guo et al., 2022). Therefore, the polarization of microglia from M1 to M2 phenotype is still used to discuss the role of microglia in AD. In the M1 state, microglia release pro-inflammatory cytokines and chemokines such as TNF-α, IL-6, IL-1β, IL-12, and CCL2. They also express iNOS, which converts arginine to NO. The accumulation of NO increases the neurotoxic effects of glutamate and damages neurons (Colonna and Butovsky, 2017). In contrast, microglia in the M2 state exhibit an anti-inflammatory phenotype, which is characterized by the production of cytokines such as IL-4, IL-10, IL-13, and TGF-β. Moreover, M2 microglia express arginase 1 to convert arginine to polyamines, which can modulate inflammation and promote tissue repair. Additionally, M2 microglia release growth factors that facilitate phagocytosis and support tissue regeneration (Zhu et al., 2019). In a state of moderate activation, microglia exhibits a dynamic balance between M1 and M2 phenotypes, enabling them to respond promptly to injurious stimuli. However, when the brain tissue is excessively stimulated, microglia undergoes a conversion toward an M1 phenotype, which is responsible for regulating inflammatory factors and causing neuronal damage. The injured neurons release toxic contents, such as Aβ, which subsequently induce microglia to adopt an M1 phenotype and release neuroimmune inflammatory factors again. This instigates a vicious cycle between damaged neurons and M1 phenotypic microglia, resulting in excessive neuroimmune inflammation and consequent neuronal death, eventually leading to brain tissue atrophy (Saito and Saido, 2018).
5.2. Inflammation mediated by microglia in AD
Inappropriate activation of immunity and inflammation is considered to be a significant contributor to the development of AD. Increased expression of markers associated with innate and adaptive immune system responses are observed in AD mouse models and patients’ brains (Zotova et al., 2013). GWS have identified risk loci that are strongly associated with the pathogenesis of AD, many of which are located near or within genes that are predominantly expressed in microglia (McQuade and Blurton-Jones, 2019). Furthermore, chronic neuroinflammation and glial cell activation have been shown to accompany AD pathology and partially mediate Aβ plaques and neurofibrillary tangles (NFTs). Risk factors for AD include pro-inflammatory gene polymorphisms, such as CCL3/MIP-1α and IL-6, which are produced by activated microglia (Zhao et al., 2018). Early microglia activation is associated with an increase in Aβ load and tau protein accumulation. However, when the Aβ load reaches a plateau, microglia activity subsequently decreases (Fan et al., 2017; Ismail et al., 2020).
In the early stages of AD, TREM2-dependent microglia activity slows down Aβ plaque formation (Parhizkar et al., 2019). Lots of activated microglia are clustered around the neuroinflammatory plaques in the brains of AD patients (Saito and Saido, 2018). M2 phenotype microglia are predominantly activated and play a role in inhibiting Aβ deposition and NFTs aggregation (Kulkarni et al., 2022). As neuritic plaques and NFTs increase, Aβ and p-tau deposits activate microglia receptors and trigger neuroimmune inflammatory responses, which exacerbate degeneration and neuronal death, leading to cognitive impairment and dementia (Maccioni et al., 2018). The immunogenicity of LPS and its highly pro-inflammatory effect on human neurons have been well established. In fact, elevated levels of LPS have been detected in the neocortex and hippocampus of brains affected by AD (Zhao et al., 2017a,b). Continuous aggregation of LPS-induced neuroimmune inflammation can lead to tau hyperphosphorylation and NFT formation, possibly through an increase in γ-secretase that results in a large accumulation of Aβ (Anthony et al., 2006). Activation of transmembrane protein receptors expressed in microglia further induces classical microglia-mediated innate immune and inflammatory responses in response to LPS (Zhao et al., 2017a,b). Finally, oxidative stress is closely related to the pathogenesis of AD (Di Bona et al., 2010). Damaging reactive oxygen species (ROS) levels are significantly higher in the AD brain than in healthy brains (Di Bona et al., 2010; Lane et al., 2018). Oxidative stress plays a significant role in the pathogenesis of AD by promoting microglia activation and morphological changes (Peters et al., 2018).
Various studies have demonstrated that microglia express different receptors, including CD36, TLR2, and TLR4, that interact with Aβ and trigger pro-inflammatory effects, resulting in chronic inflammation (Heneka et al., 2015; Su et al., 2016). In addition, microglia can activate inflammasome-related signaling upon Aβ binding, resulting in the activation of IL-1β and IL-18 through the NLR family pyrin domain containing 3 (NLRP3)/apoptosis-associated speck-like protein containing a CARD (ASC)/Caspase1 pathway, thereby contributing to the pathological progression of AD via detrimental inflammatory responses (Yang Y. et al., 2019; Zhao et al., 2020b). The β-Nicotinamide adenine dinucleotide phosphate oxidase (NOX) enzyme family, which produces ROS, is known to play a critical role in AD. Superoxide production, which is responsible for neuronal damage and initiates neuroinflammation-mediated progressive neuron degeneration, is attributed to NOX2. Pathological conditions lead to inflammation, which further activates NOX2 in microglia, eventually causing neuronal death (Siegel et al., 2019). Activation of NOX2 can lead to the production of superoxide, which in turn can activate NF-κB via various signal transduction pathways. This activation can result in the production of a large number of inflammatory factors, which may contribute to the observed neuronal death in AD (Haslund-Vinding et al., 2017; Sun et al., 2021). In summary, future therapeutic approaches for AD should also focus on reducing neuroinflammation and oxidative stress, targeting microglia activation and regulating the immune response.
6. Microglia and neuronal function
6.1. Neuronal activity
Microglia play critical roles in maintaining the integrity and plasticity of neuronal circuits in the brain. They regulate synaptic pruning and plasticity by identifying and eliminating less active synapses, preserving the strong ones (Tremblay et al., 2010; Pan and Monje, 2020). This activity is dependent on the communication between neurons and microglia. Resting microglia express the purinergic receptor, P2RY12, which enables them to respond to adenosine triphosphate (ATP) released by activated neurons. ATP functions as a chemoattractant for microglial processes, promoting communication between neurons and microglia (Peng et al., 2019; Badimon et al., 2020). Microglia break down ATP using CD39 and CD73 enzymes to produce adenosine (ADO), which then acts on the adenosine receptor, A1R, located on active neurons, thereby preventing neuronal hyperactivation (Badimon et al., 2020). This ATP-AMP-ADO-A1R-dependent feedback mechanism plays a critical role in regulating neuronal activity by preventing excessive hyperactivity that can lead to neuronal death and synaptic loss, reducing plasticity (Lepeta et al., 2016; Badimon et al., 2020). Furthermore, microglia-mediated suppression can reduce seizure severity by detecting excessive neuron excitability and decreasing neuronal firing rates.
Microglia’s frequent contact with synapses can also promote local network synchronization, followed by an increase in neuronal activity (Akiyoshi et al., 2018). Neuronal synapse activity increases when in contact with microglia, which suggests that microglia regulate neuronal activity and synchronization. In vivo two-photon Ca2+ imaging demonstrates that the synchronization of L2/3 neurons located in close proximity to one another is reduced when the normal function of microglia is disrupted, indicating that microglia can regulate neuronal activity and synchronization (Akiyoshi et al., 2018). In summary, microglia-mediated negative feedback mechanisms contribute to the homeostasis of neuronal activity, maintaining synaptic integrity and plasticity by modulating neuronal activity. Microglia play critical roles in preventing neuron hyperactivation via a negative feedback mechanism, reducing seizure severity, and promoting local network synchronization, providing a plausible explanation for how alterations in immune states may change synaptic plasticity.
6.2. Synaptic plasticity
Microglia plays a vital role in regulating synaptic plasticity. These cells constantly undergo structural changes and remodel neural circuits, contributing to the stability of LTP- the cellular mechanism of learning and memory (Poo et al., 2016; Lisman et al., 2018). The synaptic tagging and capture (STC) hypothesis suggests that weak stimulation can lead to the formation of short-lasting “synaptic tags” on certain synapses, which can be transformed into long-lasting LTP by subsequent strong stimulation from another pathway within a specific period (Frey and Morris, 1998; Nomoto and Inokuchi, 2018; Okuda et al., 2021). Under normal conditions, resting microglia promote long-term plasticity and STC. During the early phase of LTP induction, microglia set the synaptic tags that promote the stability of LTP. Eliminating microglia during this phase prevents the expression of late LTP, indicating the critical role of microglia in early STC and LTP induction (Raghuraman et al., 2019). However, once LTP is established, microglia are no longer necessary for maintaining plasticity and STC. Microglia also play a role in long-term depression (LTD), which involves the weakening of synaptic transmission. Fractalkine signaling between microglia and neurons may be involved in LTD induction. Reduced levels of fractalkine in a mouse model of Huntington’s disease result in striatal synaptic impairment and deficits in LTD induction (Kim et al., 2020). Inhibition of microglial activation prevents LTD induction, but administration of fractalkine can restore LTD (Kim et al., 2020).
The balance between proinflammatory and anti-inflammatory markers released by microglia is crucial for proper synaptic plasticity in the healthy brain (Golia et al., 2019). Disruption of this balance can lead to altered expression of inflammatory markers, resulting in decreased brain-derived neurotrophic factor (BDNF) levels and impaired phosphorylation of the AMPA receptor subunit GluR1 (Golia et al., 2019; Lin et al., 2019). The effects of proinflammatory proteins released by microglia on LTP induction appear to be transient, and the specific molecule released by microglia determines whether LTP is strengthened or impaired. For example, while overexpression of TNF-α leads to a brief increase in LTP following electrical stimulation, spatial memory deficits have been observed in TNF-α-Tg rats (Pettigrew et al., 2016). In contrast, IL-1β impairs NMDA receptor-dependent LTP at Schaffer collateral-CA1 synapses in the hippocampus (Hoshino et al., 2017). Microglia plays an essential role in regulating synaptic plasticity through their ability to release proinflammatory and anti-inflammatory mediators. The CD200/CD200R pathway and PI3K/BDNF signaling are two pathways that can modulate microglial activity and impact synaptic plasticity (Chen et al., 2017; Burk et al., 2018; Rauti et al., 2020; Saw et al., 2020). The CD200/CD200R pathway inhibits microglial activation and the release of proinflammatory mediators, promoting dendritic density and enhancing synaptic plasticity. Microglial PI3K/BDNF signaling contributes to synaptic plasticity by regulating BDNF expression, which promotes axonal branching, dendritic growth, and synapse refinement (Jung et al., 2017; Li et al., 2020; Sun et al., 2020). The balance between these molecules, including PI3K, BDNF, CREB, CD200, TNF-α, and IL-1β, determines the promotion or inhibition of synaptic plasticity. In summary, microglia plays a critical role in regulating synaptic plasticity through their ability to release proinflammatory and anti-inflammatory molecules. Resting microglia promote early STC and LTP induction, while their CD200/CD200R pathway and PI3K/BDNF signaling modulate microglial activity and impact synaptic plasticity. The specific molecule released by microglia determines whether LTP is strengthened or impaired, and the balance between proinflammatory and anti-inflammatory markers is crucial for proper synaptic plasticity in the healthy brain.
6.3. Aβ-dependent synapse loss
In AD mouse models, C1q and C3 are highly upregulated, potentially exacerbating Aβ deposition and leading to synapse loss (Naito et al., 2012; Stephan et al., 2013). Studies have shown that the depletion of C1q in AD mouse models leads to reduced microglia activation and synapse loss (Fonseca et al., 2004b; Hong et al., 2016). Similarly, in a mouse model of AD lacking C3, there was reduced synapse loss and improved cognition despite an increased Aβ load (Shi et al., 2017). This suggests that complement-mediated mechanisms play a crucial role in synapse loss and cognitive impairment in AD. One possible mechanism for synapse loss in AD involves the oligomeric form of Aβ. Aβ oligomers increase the expression of the complement protein C3 in microglia, which marks synapses and promotes microglial recruitment, leading to synapse elimination (Shi et al., 2017). Injection of Aβ oligomers in mice results in upregulation of C3 levels and microglial removal of synaptic connections (Hong et al., 2016). C3 deficiency reduces phagocytic activity and protects synapses from removal, but the involvement of other complement pathway-associated molecules in AD suggests the entire complement cascade is implicated (Czirr et al., 2017; Shi et al., 2017; Zorzetto et al., 2017). The uptake of fibrillar Aβ is mediated through the C3 receptor (CD11b) and the receptor for C5a, which is upregulated around plaques and correlates with Aβ and neurofibrillary pathology (Ager et al., 2010; Fonseca et al., 2013). Inhibiting the C5aR pathway reduces Aβ pathology, neurofibrillary tangles, and improves behavior in AD mouse models (Fonseca et al., 2009). Aβ oligomers also selectively target dendritic spines, as evidenced by their colocalization with PSD-95, a marker of these structures (Lacor et al., 2004). Mechanistically, Aβ binds to glutamatergic receptors at the postsynaptic site leading to their inactivation and could act as an extracellular hook to recruit microglia to the synapse, inducing removal of the Aβ-tagged synapse (Decker et al., 2010; Li et al., 2011). One could also envision that fibrillar Aβ first recruits complement molecules, which in turn promote microglial recruitment (Eikelenboom and Veerhuis, 1996; Stoltzner et al., 2000). Because C3 is a known mediator for microglial phagocytosis of fibrillar Aβ, Aβ oligomer deposition at the synapse could induce not only microglial recruitment but also, via upregulation of C3, promote engulfment of synaptic structures (Maier et al., 2008; Fu et al., 2012; Figure 6).
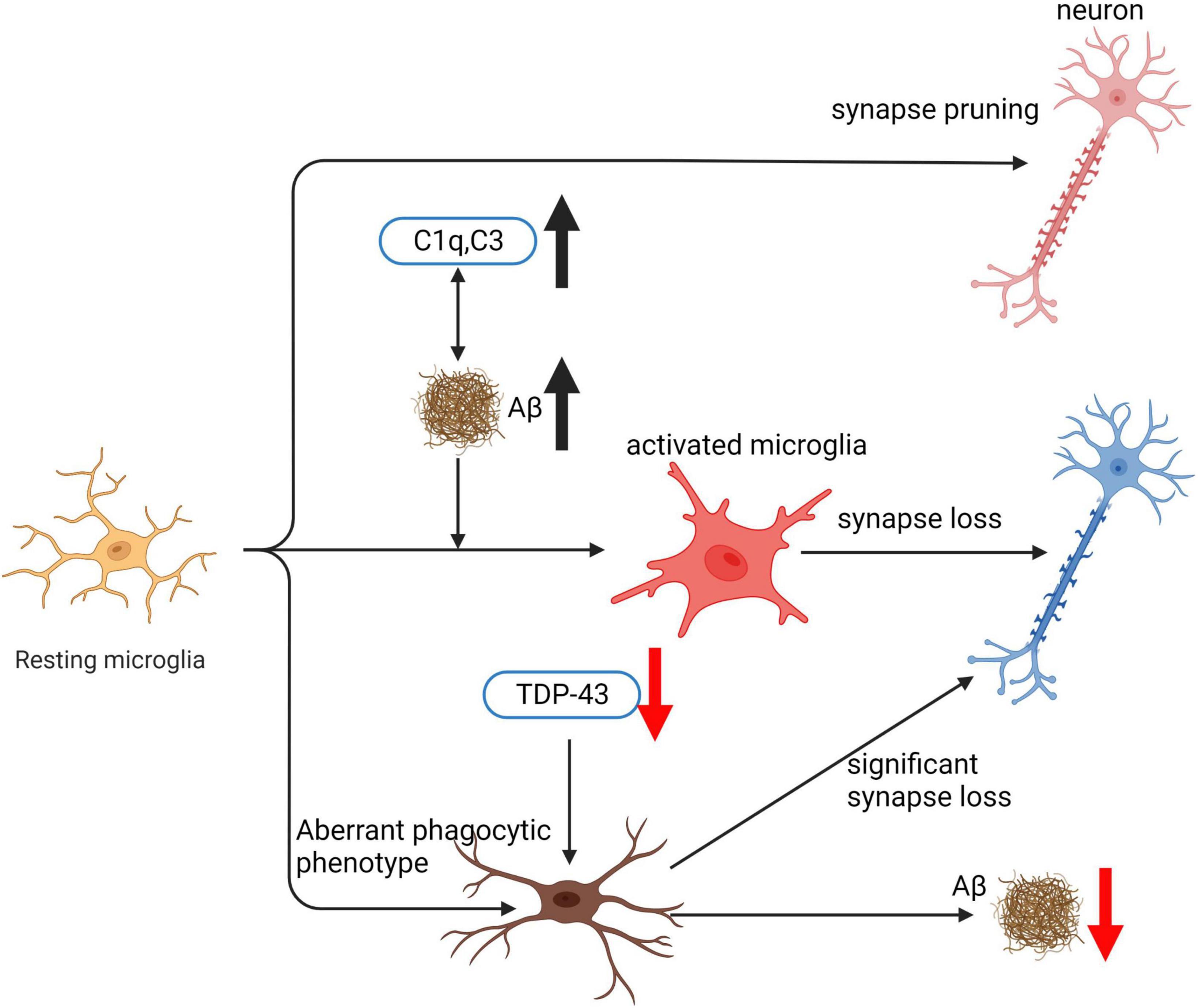
Figure 6. Mechanisms of synaptic loss induced by microglia under normal and pathological conditions. This illustration depicts the various pathways through which microglia contribute to neuronal synaptic loss. Under normal circumstances, microglia participate in routine synapse pruning. However, upon aggregation of Aβ, levels of complement proteins C1q and C3 rise, which in turn promote further Aβ aggregation. This initiates a vicious cycle leading to the activation of microglia, resulting in synaptic loss in neurons. Moreover, the knockout of TDP-43 causes an aberrant phagocytic phenotype in neurons, enhancing their phagocytic function. While this increase in phagocytic activity promotes Aβ clearance, it also triggers a substantial loss of synapses in the neurons.
6.4. Aβ-independent synapse loss
During early development, microglia play a critical role in synapse pruning (Paolicelli et al., 2011; Schafer et al., 2012), suggests that microglia’s physiological role in synapse pruning occurs well before the onset of aging or Aβ deposition. Therefore, it is plausible that the reactivation of this function during aging may not solely depend on Aβ. Selectively depleting TDP-43 in a microglial cell line led to an aberrant phagocytic phenotype, which resulted in enhanced Aβ clearance (Paolicelli et al., 2017). Notably, mice expressing mutant APP and lacking microglial TDP-43 displayed enhanced Aβ clearance, but at the same time, they exhibited significant synapse loss. Remarkably, the deposition of Aβ is not a prerequisite for this synaptic loss, as mice lacking any human mutant APP also exhibited a similar loss of synapses (Paolicelli et al., 2017). Another study demonstrated that 16-month-old AD mice deficient in complement C3 exhibited less synaptic loss and performed better in behavioral tests of memory, despite an increase in Aβ load (Shi et al., 2017). These findings suggest that the trajectory of synapse loss may not necessarily follow that of Aβ buildup. Taken together, these studies provide compelling evidence that microglial dysfunction, independent of Aβ pathology, may contribute to the loss of synapses. Such findings may have significant implications for the development of therapeutic strategies aimed at preventing or delaying synaptic loss in neurodegenerative diseases (Figure 6).
6.5. Learning and memory
Microglia have been extensively studied for their role in learning and memory. Previous research suggested a negative impact of microglia on these processes. However, recent studies have provided insights into the specific mechanisms through which microglia enhance memory by regulating synaptic pruning and neuronal remodeling. This suggests that microglia play a positive role in memory formation and consolidation (Mohammadi et al., 2020; Worthen et al., 2020; Liao et al., 2021). Microglia play a vital role in regulating the quality of memory by removing the ECM, which is involved in neuronal plasticity, learning, and memory (Vainchtein et al., 2018). This process is facilitated by the signaling of a cytokine called IL-33, which activates microglia to promote the formation of new spines and regulate neuronal plasticity in response to experiences. In mice, a loss of IL-33 receptors on microglia or neuronal IL-33 expression results in impaired synaptic plasticity and reduced precision of fear memory (Nguyen et al., 2020). Moreover, studies using transgenic mice have demonstrated the importance of IL-33 signaling in regulating memory quality. For example, IL-33 knockout mice exhibited impaired fear memory precision and reduced newborn neuron integration (Nguyen et al., 2020). Acute microglia depletion has also been found to prevent forgetting induced by pro-neurogenic drugs, highlighting the role of microglia in regulating forgetting of remote memories (Wang C. et al., 2020). Interestingly, treatment with IL-33 has been shown to improve cognitive deficits and synaptic plasticity in AD mice (Fu A. K. Y. et al., 2016). IL-33 has been found to enhance the recruitment of microglia and their interaction with plaques, resulting in increased phagocytic activity and a reduction in the size of plaques. Moreover, IL-33 administration has been shown to mitigate inflammation and upregulate anti-inflammatory genes in the brain. Additionally, microglia are involved in the process of forgetting by actively participating in adult neurogenesis through phagocytosis (Bennett and Molofsky, 2019; Wang C. et al., 2020). They identify and remove most of the newborn neurons that undergo apoptosis after formation (Diaz-Aparicio et al., 2020). When the phagocytic function of microglia is impaired for an extended period, it results in a decrease in adult hippocampal neurogenesis. Conversely, when there is a short-term deficiency in phagocytic function, it transiently enhances neurogenesis in the same area (Diaz-Aparicio et al., 2020). The morphology and function of microglia play critical roles in the regulation of learning and memory. Studies have shown that acute ablation of microglia does not impact short-term memory; however, repopulated microglia with an amoeboid morphology have been associated with enhanced memory and increased numbers of mature neurons (Gipson and Olive, 2017; De Luca et al., 2020). In summary, the studies on microglia and their impact on learning and memory underscore their intricate and fluid involvement in these processes. Microglia perform diverse functions, such as controlling memory quality through the IL-33 signaling pathway, regulating forgetting via phagocytosis and complement pathways, and influencing memory-related processes through their morphology and activity. These findings establish the indispensable role of microglia in learning and memory.
7. Therapeutic approaches
7.1. Therapeutic approaches based on inhibiting microglia function
7.1.1. Microglial depletion
Multiple research studies have underscored the significance of specific subsets of microglia, particularly disease-associated microglia (DAM), in the progression of AD. The conjecture underpinning microglial depletion is that certain activated microglia, including DAM, may accentuate AD pathogenesis through a series of actions, such as facilitating the accumulation of Aβ plaques, amplifying neuroinflammation, and potentially influencing tau pathology (Keren-Shaul et al., 2017; Krasemann et al., 2017). DAMs, characterized by a unique transcriptional profile and often found in close proximity to amyloid plaques, are thought to have substantial impacts on neuronal health and function (Mathys et al., 2017; Deczkowska et al., 2018). Consequently, it has been proposed that therapeutic strategies should be more disease-specific and aim at selectively targeting these DAMs (Keren-Shaul et al., 2017). The implementation of microglial depletion has been achieved through various strategies, most notably using pharmacological and genetic techniques. A common method involves the use of brain-penetrating inhibitors of CSF1R, a crucial cell surface receptor for microglial survival and proliferation. In AD mouse models, these inhibitors have demonstrated improvements in cognition and reductions in both neuroinflammation and neuritic plaque formation (Dagher et al., 2015; Olmos-Alonso et al., 2016; Spangenberg et al., 2016; Sosna et al., 2018). However, the relationship between microglial depletion and amyloid pathology has been inconsistent, highlighting the need for further investigation (Casali et al., 2020). The potential of microglial depletion as a therapeutic approach must be carefully evaluated, considering the possible unforeseen consequences and the critical role microglia play in maintaining brain health (Guo et al., 2019; Oosterhof et al., 2019). Therefore, more research is warranted to further elucidate the complex roles of microglia, particularly DAM, in AD pathogenesis and treatment.
7.1.2. Inhibiting the inflammatory response in microglia
Focusing on particular pro-inflammatory pathways, rather than depleting microglia, may provide a more efficacious therapeutic strategy for AD. One such pathway is the inflammasome, which plays a crucial role in microglial activation and induces the release of pro-inflammatory cytokines, ultimately leading to neuronal damage. Targeting the inflammasome pathway represents an encouraging potential treatment strategy for AD (Heneka et al., 2018; Scheiblich et al., 2020). Several medications have been developed to target different elements of the inflammasome, including purinergic receptors, caspase-1, and NLRP3 (Thawkar and Kaur, 2019). For example, purinergic receptor antagonists can regulate extracellular ATP levels, which participate in the activation of the NLRP3 inflammasome and the subsequent release of pro-inflammatory cytokines. Caspase-1 inhibitors obstruct the activation of IL-1β and IL-18, two pivotal cytokines in the inflammasome pathway, thereby attenuating the inflammatory response. Direct inhibitors of NLRP3 can hinder its activation, oligomerization, or interaction with other inflammasome components, ultimately preventing inflammasome assembly and downstream inflammatory signaling. These therapeutic interventions hold the potential to reduce neuroinflammation, alleviate neuronal damage, and decelerate AD progression. Nevertheless, it is vital to carry out additional pre-clinical and clinical research to determine the safety and efficacy of these drugs, as well as to comprehend the potential side effects and long-term consequences of targeting specific inflammasome components. By deepening our understanding of the molecular mechanisms and interactions within the inflammasome pathway, we can continue to develop innovative and targeted therapies for AD that minimize adverse effects and maximize therapeutic potential.
7.1.2.1. Targeting purinergic receptors to inhibit microglial inflammation in AD
Emerging as potential therapeutic targets for AD, purinergic receptors, especially the P2 × 7R subtype found on microglia, play dual roles in neuroprotection and neurodegeneration (Burnstock, 2016; Woods et al., 2016). P2 × 7R antagonism may offer significant therapeutic benefits for addressing neuroinflammation and other neurodegenerative conditions, as it occurs exclusively under high ATP concentrations, which are believed to increase during the progression of neuroinflammatory diseases (Sperlágh and Illes, 2014; Bhattacharya, 2018; Burnstock and Knight, 2018). Research has highlighted the importance of P2 × 7R in the development of neurodegenerative disorders through both in vitro and in vivo studies. In the context of AD, characterized by reactive microgl lia and Aβ plaques, Aβ is known to stimulate neuroinflammation via P2 × 7R modulation (Sanz et al., 2009). Targeting P2Y2R during the early stages of AD could be beneficial due to its role in tissue repair (Peterson et al., 2010). Brilliant blue G (BBG), a P2 × 7R antagonist derived from the blue food dye FD&C blue No. 1, has shown promise in treating neurological disorders due to its unique ability to cross the blood-brain barrier. Demonstrating a favorable safety profile in healthy animal models, BBG has potential clinical applications (Borzelleca et al., 1991). Widely recognized for its effectiveness on P2 × 7R in various species, BBG has been observed to enhance cognition and promote dendritic spine development in Aβ42-injected rats (Chen et al., 2014). Furthermore, BBG exhibits neuroprotective properties by counteracting the inflammatory responses induced by P2 × 7R agonists in Aβ-peptide-injected rat brains (Ryu and McLarnon, 2008). GlaxoSmithKline has developed a P2 × 7 receptor antagonist, GSK1482160, which exhibits superior CNS penetration and the ability to be radiolabeled with 11C. The radio labeled 11C-GSK1482160 displays high P2 × 7 selectivity and functions as a neuroinflammation biomarker in LPS-challenged rats (Territo et al., 2017).
7.1.2.2. Inhibiting NLRP3 inflammasome activation
Several compounds have demonstrated potential in modulating the NLRP3/caspase-1 inflammasome pathway in AD. Pterostilbene, for instance, has been found to be effective in reducing inflammasome activation and lowering the secretion of proinflammatory mediators in BV-2 microglia at a concentration of 10 μM (Li J.-J. et al., 2018; Li Q. et al., 2018). Similarly, JC-124, a small molecule NLRP3 inflammasome inhibitor, led to reduced levels of Aβ deposition and decreased levels of soluble and insoluble Aβ1–42 in the brain of TgCRND8 mice (Yin et al., 2018). MCC950, a NLRP3 inhibitor, has been found to prohibit inflammasome activation and IL-1β release from microglia. Moreover, it promoted Aβ phagocytosis in the APP/PS1 mouse model of AD (Dempsey et al., 2017). In addition, Benzyl isothiocyanate (BITC), a naturally occurring compound found in cruciferous vegetables, has been found to inhibit the secretion of IL-1β, inflammasome activation, and proinflammatory mediators in BV2 microglial cells (Lee et al., 2016). Furthermore, Stavudine, a nucleoside reverse transcriptase inhibitor, has been found to downregulate NLRP3 inflammasome activation and Aβ autophagy in cell cultures of THP-1-derived macrophages induced by Aβ (La Rosa et al., 2019). Similarly, Artemisinin, obtained from Artemisia annua and well-known for its antimalarial effect, has been found effective in treating AD by inhibiting activation of NF-κB and NLRP3 inflammasome in 5-month-old APPswe/PS1dE9 transgenic mice (Shi et al., 2013). Finally, Dihydromyricetin (DHM) has been found to decrease activation of the NLRP3 inflammasome and reduce memory and cognition deficits in APP/PS1 double-transgenic mice (Feng et al., 2018). These findings suggest that targeting the NLRP3 inflammasome may offer a promising therapeutic strategy for the treatment of AD.
7.1.2.3. Caspase-1 inhibition: a potential strategy for modulating microglial activation in AD
Caspase 1 has gained attention as a potential therapeutic target for addressing age-related cognitive decline and AD. Bacopa, a naturally occurring compound, exhibits inhibitory effects on several enzymes involved in neuroinflammation and neurodegeneration, including Matrix Metalloproteinase-3 (MMP-3), caspase 1, and caspase 3. In a cell-free assay, Nemetchek et al. (2017) showed that Bacopa effectively suppressed the activity of these enzymes, supporting its potential as a therapeutic agent for AD. Additionally, VX-765, a selective caspase-1 inhibitor, has demonstrated promising effects in AD mouse models. It has been found to attenuate Aβ peptide accumulation, mitigate brain inflammation, and preserve normal synaptophysin protein levels in the mouse hippocampus. These outcomes were dose-dependent and observed in J20 mice (Flores et al., 2018). Other studies have also reported the neuroprotective effects of caspase-1 inhibition, further emphasizing its potential as a therapeutic strategy for AD (Kaushal et al., 2015).
7.1.2.4. RIPK1 inhibition: a pathway to mitigate microglial-driven pathology in AD
The role of receptor-interacting serine/threonine kinase 1 (RIPK1) activation in microglia is thought to be crucial in the development of AD. While RIPK1 has been linked to the inflammasome response, recent research indicates that its function in this process is primarily independent of its kinase activity (Malireddi et al., 2020). Notably, increased RIPK1 kinase activity has been observed in animal models of AD, amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS), as well as in human patient samples, implying a potential contribution of this protein to the pathogenesis of these neurodegenerative disorders (Yuan et al., 2019). Various pre-clinical studies have underscored the potential protective effects of utilizing either a kinase-dead mutant of RIPK1 (RIPK1D138N) or pharmacological RIPK1 inhibitors in diverse neurodegenerative disease models. RIPK1 activity is known to modulate cell death processes such as apoptosis and necroptosis and to regulate the production of inflammatory cytokines in specific cell types, including microglia (Degterev et al., 2019; Yuan et al., 2019). Intriguingly, RIPK1 activity has also been demonstrated to regulate metabolic adaptation and reactive oxygen species production in response to TNF-a, which may further contribute to pathological microglial cell states (Zhang et al., 2009). Owing to these findings, ongoing clinical trials are exploring the use of small-molecule RIPK1 inhibitors for AD. Collectively, these studies suggest that targeting RIPK1 may offer therapeutic potential in the management of neurodegenerative diseases.
7.2. Therapeutic approaches based on enhancing microglia function
7.2.1. Exploring TREM2: modulating microglial activity as a therapeutic strategy for AD
Although TREM2-dependent activities may not significantly reduce amyloid plaque load in mouse models, they can promote plaque compaction and alleviate plaque-associated neuritic pathology (Ulrich et al., 2014; Wang et al., 2016; Meilandt et al., 2020). As a result, TREM2 has emerged as a primary therapeutic target for AD, with researchers exploring methods for selective modulation to achieve favorable outcomes. Disease-associated TREM2 variants can result in loss of function in mouse and cellular models, implying that enhancing TREM2 function could be beneficial (Heslegrave et al., 2016; Piccio et al., 2016; Suárez-Calvet et al., 2019). Indirect evidence from AD patients suggests that higher TREM2 levels may have protective effects on disease progression. Several independent cohorts have reported increased levels of soluble TREM2 (sTREM2) concentrations in cerebrospinal fluid (CSF) early in the disease, which could serve as a potential marker for full-length TREM2-mediated signaling. Elevated sTREM2 levels have been detected in patients with mild cognitive impairment and dominant inherited AD, up to 5 years before the estimated year of onset (Suárez-Calvet et al., 2016a). However, no correlation was found between sTREM2 and amyloid deposition, suggesting that TREM2 expression might increase in response to tau-related cellular pathology. Research has also found that sTREM2 levels rise during physiological aging in humans and mice, with TREM2-dependent increases in microglial nodules observed during aging in mice (Suárez-Calvet et al., 2016a; Kleinberger et al., 2017). It is hypothesized that these microglial accumulations are a response to myelin damage, supported by studies in demyelination models (Poliani et al., 2015; Nugent et al., 2020). Additionally, in humans, a higher ratio of CSF sTREM2 over phospho-tau predicted slower clinical progression and was associated with slower cognitive decline (Ewers et al., 2019). The cumulative evidence suggests that TREM2 plays a beneficial role in the CNS, which could be amplified to prevent or slow the progression of sporadic AD. Agonist antibodies binding to the extracellular region and initiating receptor signaling may hold potential for AD treatment (Figure 7; Cignarella et al., 2020; Price et al., 2020; Schlepckow et al., 2020; Wang S. et al., 2020). Modulating TREM2 activity represents a promising avenue for developing effective therapeutic interventions for AD. Mouse TREM2 studies have shown that antibody stimulation enhances microglial chemotaxis, clustering around Aβ plaques, and phagocytosis of cellular debris, possibly facilitating Aβ plaque removal (Wang et al., 2016; Mazaheri et al., 2017; Parhizkar et al., 2019). However, research on an anti-mouse TREM2 antibody that competes for ligand binding and inhibits cell autonomous signaling revealed that TREM2 antagonism significantly worsened the outcome in experimental autoimmune encephalomyelitis (EAE). Currently, three promising strategies for modulating TREM2 activity have emerged. Two companies have identified antibodies that enhance TREM2 signaling through crosslinking (Cheng et al., 2018; Cignarella et al., 2020; Price et al., 2020; Wang S. et al., 2020), while the third approach aims to prevent shedding of the extracellular domain by ADAM family proteases (Wunderlich et al., 2013; Schlepckow et al., 2020). However, selectively inhibiting TREM2 shedding is preferred over inhibiting ADAM10, which cleaves other vital physiological substrates in the brain (Kuhn et al., 2016; Schlepckow et al., 2020). One promising antibody clone, 4D9, binds to an epitope only 12 amino acids N-terminal to the TREM2 protein, effectively obstructing the shedding of soluble TREM2. In vitro biochemical assays have demonstrated that 4D9 can impede the cleavage of the TREM2 stalk peptide, activating DAP12/phosphorylation/Syk signaling, which is crucial for downstream signaling (Cignarella et al., 2020; Price et al., 2020; Wang S. et al., 2020). Investigations have revealed that 4D9 efficiently binds sTREM2 in the CSF when administered at high doses peripherally, suggesting that it can engage the target in the CNS in vivo (Wang S. et al., 2020). Studies have assessed the effects of AL002a/c, a humanized monoclonal IgG1 antibody that binds to TREM2 and activates the TREM2 signaling pathway, and 4D9 on Aβ accumulation in transgenic AD mouse models (Price et al., 2020; Schlepckow et al., 2020; Wang S. et al., 2020). Surprisingly, neither AL002c nor 4D9 enhanced the clustering of microglia around amyloid plaques, while AL002a did. In contrast, AL002a significantly reduced the total Aβ plaque load in the same model and decreased fibrillar Aβ within the hippocampus. Furthermore, both AL002a/c improved behavior in 5 × FAD mice and reduced plasma neurofilament light chain, a marker for neurodegeneration (Wang S. et al., 2020). In summary, the AL002 family of antibodies shares numerous properties, but they display divergent activities toward Aβ clearance and microglial clustering. The reasons for these discrepancies remain unclear but could stem from differences in mouse models or dosing paradigms utilized in these studies.
Figure 7. Outlines therapeutic strategies targeting microglial lipid and lysosomal functions. Phagocytic activity and apolipoprotein uptake, such as APOE, are crucial for delivering lipids to endolysosomal compartments for degradation. During this process, cholesterol is liberated from digested phagocytic cargo, such as apoptotic cells. Subsequently, cholesterol can exit the endolysosomal compartment and be esterified into smaller lipid molecules. These lipids can either be stored in lipid droplets or exported from the cell via lipid transporters. TREM2 dysfunction can result in various microglial defects, including diminished phagocytosis, impaired lysosomal degradation, and compromised cholesterol efflux, as well as the accumulation of neutral lipids in lipid droplets. Multiple therapeutic opportunities exist for targeting microglial function in AD. TREM2-targeting antibodies can enhance phagocytosis and lysosomal function. Small molecules targeting ACAT can reduce lipid droplet formation, while various nuclear receptors can promote cholesterol efflux. Additionally, lysosomal function activation can be achieved through small molecules that induce lysosomal acidification or by stimulating the activation of TFEB family members to enhance lysosomal function.
7.2.2. Enhancing lysosomal activity
Microglial phagocytosis is a multifaceted process that necessitates substantial alterations in microglial gene expression and a robust lysosomal degradative capacity. Microglia express high levels of genes controlling lysosomal function, including hydrolases and regulators such as cathepsins, lipases, prosaposin, and progranulin. However, AD may impair lysosomal degradative capacity. For example, Aβ aggregates can undermine microglial endolysosomal membrane integrity, potentially disrupting lysosomal compartment proton gradients and releasing cathepsins into the cytosol (Heneka et al., 2018). Additionally, lysosomal dysfunction is evident in aged murine microglia, characterized by lipofuscinosis (Safaiyan et al., 2016). Enhancing lysosomal function might be a viable therapeutic approach to improve microglial function (Figure 7). Targeting lysosomal pH control, such as increasing the stability or activity of ClC-7 in microglia or promoting V-type ATPase activity, may facilitate the degradation of age- and disease-associated phagocytic cargoes (Majumdar et al., 2011). An alternative strategy to boost lysosomal biogenesis involves enhancing signaling pathways controlling this process at the transcriptional level. Key regulators of genes encoding hydrolases, membrane proteins, proton pumps, and macroautophagy factors crucial for proper lysosomal function include transcription factor EB (TFEB) and its related family members TFE3, TFEC, and MITF. These transcription factors act as master regulators, playing a critical role in ensuring lysosomal functionality (Sardiello et al., 2009). In microglia, nutrient deprivation and mTORC1 signaling downregulation or lysosomal dysfunction activate the TFEB pathway (Sardiello et al., 2009). The coordinated lysosomal expression and regulation (CLEAR) network is activated in vivo in lysosomal storage disease models, including Ids and Grn KO, though specific brain cell types might be differentially affected (Götzl et al., 2018; Ullman et al., 2020). In microglia, not only nutrient deprivation but also immune stimuli such as TLR agonists can activate TFEB and TFE3. This activation leads to the induction of various pro-inflammatory cytokines, as well as lysosomal and autophagic genes. While the activation of the TFEB/TFE3 pathway may promote lysosomal degradation and autophagy, it could also enhance inflammatory responses, resulting in either beneficial or detrimental outcomes depending on the disease or disease stage (Götzl et al., 2018; Ullman et al., 2020). From a therapeutic perspective, exploiting endogenous regulators of the TFEB pathway in microglia, such as mTORC1 pathway antagonists, may be a potential approach (Puertollano et al., 2018). Enhancing lysosomal biogenesis in microglia could help clear disease-associated lysosomal cargoes, including protein aggregates like Aβ and tau, as well as lipid-rich cellular debris.
7.2.3. Enhancing lipid processing
The study of LOAD genetics has pinpointed lipid metabolism as a crucial risk factor (Efthymiou and Goate, 2017). The discovery of APOE4 as the primary genetic susceptibility gene for AD has raised the possibility that lipid transport, including cholesterol and triglyceride metabolism, might be implicated in the disease mechanism (Corder et al., 1993). It has been demonstrated that APOE isoforms differentially impact peripheral lipid metabolism (Leduc et al., 2010; Zhao et al., 2020c), and subsequent investigations have concentrated on the relationship between APOE and Aβ deposition in the brain parenchyma and amyloid efflux from the brain (Huynh et al., 2017). Recent findings suggest that the lipidation degree of APOE may influence amyloidogenesis, as poorly lipidated APOE4 has shown a higher propensity for both aggregation and Aβ seeding (Huynh et al., 2017). Additionally, microglial APOE has been found to play a critical role in immunometabolism, with dysfunction in these cells leading to two distinct types of cholesterol-processing impairments: the accumulation of lysosomal cholesterol and cholesterol crystals, as well as the buildup of cholesteryl esters, the stored form of cholesterol, within lipid droplets (Cantuti-Castelvetri et al., 2018; Nugent et al., 2020). Cholesteryl ester accumulation has been observed in microglia that lack either TREM2 or its signaling component, phospholipase Cγ2, encoded by the LOAD-associated PLCG2 gene (Andreone et al., 2020; Nugent et al., 2020). This observation aligns with prior lipidomic analyses of LOAD patient brain samples and 5 × FAD mouse models (Chan et al., 2012). Furthermore, studies involving APOE knockout microglia and TREM2 knockout induced microglia have highlighted the indispensable role of apolipoprotein in central nervous system cholesterol transport during demyelination events. These investigations emphasize the importance of understanding lipid metabolism in the context of neuroinflammation and neurodegeneration (Cantuti-Castelvetri et al., 2018; Andreone et al., 2020; Nugent et al., 2020). The presence of lipid droplets containing cholesteryl esters and cholesterol in lysosomes leads to the “foamy” appearance of diseased microglia, which can cause lipotoxicity and cellular stress (Moore and Tabas, 2011; Olzmann and Carvalho, 2019). This can contribute to decreased cell survival, heightened pro-inflammatory signaling, oxidative stress, and inflammasome activation (Moore and Tabas, 2011). To address these issues, nuclear receptors, such as PPARs, LXRs, and RXRs, have been targeted as potential therapeutics to promote lipid efflux while reducing inflammation (Moutinho and Landreth, 2017). Although some agonists have shown efficacy in AD mouse models, such as reducing Aβ burden and exerting anti-inflammatory actions in the brain, their clinical efficacy remains uncertain (Moutinho and Landreth, 2017). For example, LXR agonists, such as GW3965, have demonstrated profound effects in microglia by rescuing cholesterol and cholesteryl ester accumulation in APOE KO microglia and TREM2 KO iMG (Cantuti-Castelvetri et al., 2018; Nugent et al., 2020). Furthermore, targeting ACAT1, the enzyme that catalyzes the esterification of cholesterol, through inhibition or ablation, has demonstrated the potential to decrease Aβ and tau pathology in mouse models of AD. This effect is believed to be mediated through modulation of both APP processing and autophagy (Puglielli et al., 2001; Hutter-Paier et al., 2004; Shibuya et al., 2015). Reducing cholesteryl esters may decrease oxidized cholesteryl ester species and minimize the deleterious effects of lipid peroxidation (Nugent et al., 2020). However, reducing cholesterol esterification may increase cellular levels of free cholesterol, which requires efficient efflux mechanisms for its elimination from the cells. In conclusion, understanding the critical role of lipid metabolism in LOAD and identifying potential therapeutic targets may lead to more effective treatments for this debilitating disease (Figure 7).
8. Summary and outlook
In this review, we have provided a comprehensive overview of the critical role microglia play in AD, covering key aspects such as the associated genetic mutations, microglial phenotypes, phagocytic functions related to Aβ and apoptotic cells, the involvement of inflammatory pathways, and the relationship between microglia, synaptic loss, and synaptic plasticity. Moreover, we have highlighted the development of AD-related therapeutics targeting microglia and the current limitations in our understanding of microglial biology.
In conclusion, it is evident that microglia are crucial players in the pathogenesis of AD, and a deeper understanding of their diverse functions and interactions with other cellular components in the brain will be vital for the development of effective therapeutic strategies. However, despite considerable progress in recent years, there remain significant gaps in our knowledge of microglial biology, particularly concerning their heterogeneity, precise mechanisms of action, and the interplay between various signaling pathways.
Future research should continue to unravel the complexity of microglial biology, explore the underlying molecular mechanisms that drive their diverse functions, and investigate the potential of modulating microglial activity to halt or reverse disease progression. Additionally, the development of novel in vitro and in vivo models that more accurately recapitulate the human disease will be crucial for the validation of new therapeutic targets and for testing candidate drugs. Advances in single-cell sequencing technologies and systems biology approaches will undoubtedly contribute to a better understanding of microglial heterogeneity and their functional STATes in AD.
One notable limitation of current research is the lack of standardized criteria for defining microglial phenotypes and functional states, which may lead to inconsistencies in the literature and hinder the development of targeted therapies. Furthermore, many studies focus on individual aspects of microglial biology, which may not fully capture the complex interplay between various microglial functions in the context of AD.
The design and implementation of clinical trials that specifically target microglial pathways should consider factors such as disease stage, genetic background, and individual patient characteristics to achieve optimal therapeutic outcomes. Moreover, addressing the current limitations in our understanding of microglial biology and function will be essential to harness the potential of microglia-targeted therapies for the prevention, management, and eventual cure of AD. Together, these efforts will pave the way for a new era of precision medicine, capitalizing on the promise of microglia-targeted interventions for AD.
Author contributions
JM conceptualized the study, performed the literature search, and drafted the manuscript. YY, HM, and YL were responsible for critically reviewing and analyzing the literature. JZ, CL, and MY contributed to data interpretation and provided expert input. JL supervised the overall project, provided critical feedback, and revised the manuscript for important intellectual content. All authors contributed to the final version of the manuscript and approved it for submission.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Afagh, A., Cummings, B. J., Cribbs, D. H., Cotman, C. W., and Tenner, A. J. (1996). Localization and cell association of C1q in Alzheimer’s disease brain. Exp. Neurol. 138, 22–32. doi: 10.1006/exnr.1996.0043
Ager, R. R., Fonseca, M. I., Chu, S.-H., Sanderson, S. D., Taylor, S. M., Woodruff, T. M., et al. (2010). Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer’s disease. J. Neurochem. 113, 389–401. doi: 10.1111/j.1471-4159.2010.06595.x
Ahmadian, M., Suh, J. M., Hah, N., Liddle, C., Atkins, A. R., Downes, M., et al. (2013). PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med. 19, 557–566. doi: 10.1038/nm.3159
Akimov, S. S., Krylov, D., Fleischman, L. F., and Belkin, A. M. (2000). Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 148, 825–838. doi: 10.1083/jcb.148.4.825
Akiyoshi, R., Wake, H., Kato, D., Horiuchi, H., Ono, R., Ikegami, A., et al. (2018). Microglia enhance synapse activity to promote local network synchronization. eNeuro 5:ENEURO.0088-18.2018. doi: 10.1523/ENEURO.0088-18.2018
Ando, K., Nagaraj, S., Küçükali, F., de Fisenne, M.-A., Kosa, A.-C., Doeraene, E., et al. (2022). PICALM and Alzheimer’s disease: an update and perspectives. Cells 11:3994. doi: 10.3390/cells11243994
Andreone, B. J., Przybyla, L., Llapashtica, C., Rana, A., Davis, S. S., van Lengerich, B., et al. (2020). Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat. Neurosci. 23, 927–938. doi: 10.1038/s41593-020-0650-6
Andrews-Zwilling, Y., Bien-Ly, N., Xu, Q., Li, G., Bernardo, A., Yoon, S. Y., et al. (2010). Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 30, 13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010
Anthony, I. C., Ramage, S. N., Carnie, F. W., Simmonds, P., and Bell, J. E. (2006). Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 111, 529–538. doi: 10.1007/s00401-006-0037-0
Arandjelovic, S., and Ravichandran, K. (2015). Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 16, 907–917.
Badimon, A., Strasburger, H. J., Ayata, P., Chen, X., Nair, A., Ikegami, A., et al. (2020). Negative feedback control of neuronal activity by microglia. Nature 586, 417–423. doi: 10.1038/s41586-020-2777-8
Barczyk, M., Carracedo, S., and Gullberg, D. (2010). Integrins. Cell Tissue Res. 339, 269–280. doi: 10.1007/s00441-009-0834-6
Beamer, E., Fischer, W., and Engel, T. (2017). The ATP-Gated P2X7 receptor as a target for the treatment of drug-resistant epilepsy. Front. Neurosci. 11:21. doi: 10.3389/fnins.2017.00021
Beck, T. N., Nicolas, E., Kopp, M. C., and Golemis, E. A. (2014). Adaptors for disorders of the brain? the cancer signaling proteins NEDD9, CASS4, and PTK2B in Alzheimer’s disease. Oncoscience 1, 486–503. doi: 10.18632/oncoscience.64
Beecham, G. W., Bis, J. C., Martin, E. R., Choi, S.-H., DeStefano, A. L., van Duijn, C. M., et al. (2017). The Alzheimer’s disease sequencing project: study design and sample selection. Neurol. Genet. 3:e194. doi: 10.1212/NXG.0000000000000194
Beecham, G. W., Vardarajan, B., Blue, E., Bush, W., Jaworski, J., Barral, S., et al. (2018). Rare genetic variation implicated in non-Hispanic white families with Alzheimer disease. Neurol. Genet. 4:e286. doi: 10.1212/NXG.0000000000000286
Bennett, F. C., and Molofsky, A. V. (2019). The immune system and psychiatric disease: a basic science perspective. Clin. Exp. Immunol. 197, 294–307. doi: 10.1111/cei.13334
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23. doi: 10.1038/ng1934
Bettadapur, A., Miller, H. W., and Ralston, K. S. (2020). Biting off what can be chewed: trogocytosis in health, infection, and disease. Infect. Immun. 88:e00930-19. doi: 10.1128/IAI.00930-19
Bhattacharya, A. (2018). Recent advances in CNS P2X7 physiology and pharmacology: focus on neuropsychiatric disorders. Front. Pharmacol. 9:30. doi: 10.3389/fphar.2018.00030
Bhattacherjee, A., Daskhan, G. C., Bains, A., Watson, A. E. S., Eskandari-Sedighi, G., St Laurent, C. D., et al. (2021a). Increasing phagocytosis of microglia by targeting CD33 with liposomes displaying glycan ligands. J. Control. Release 338, 680–693. doi: 10.1016/j.jconrel.2021.09.010
Bhattacherjee, A., Jung, J., Zia, S., Ho, M., Eskandari-Sedighi, G., St Laurent, C. D., et al. (2021b). The CD33 short isoform is a gain-of-function variant that enhances Aβ(1-42) phagocytosis in microglia. Mol. Neurodegener. 16:19. doi: 10.1186/s13024-021-00443-6
Bhattacherjee, A., Rodrigues, E., Jung, J., Luzentales-Simpson, M., Enterina, J. R., Galleguillos, D., et al. (2019). Repression of phagocytosis by human CD33 is not conserved with mouse CD33. Commun. Biol. 2:450. doi: 10.1038/s42003-019-0698-6
Biber, K., Neumann, H., Inoue, K., and Boddeke, H. W. G. M. (2007). Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 30, 596–602. doi: 10.1016/j.tins.2007.08.007
Bis, J. C., Jian, X., Kunkle, B. W., Chen, Y., Hamilton-Nelson, K. L., Bush, W. S., et al. (2020). Whole exome sequencing study identifies novel rare and common Alzheimer’s-associated variants involved in immune response and transcriptional regulation. Mol. Psychiatry 25, 1859–1875. doi: 10.1038/s41380-018-0112-7
Boisvert, W. A., Rose, D. M., Boullier, A., Quehenberger, O., Sydlaske, A., Johnson, K. A., et al. (2006). Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler. Thromb. Vasc. Biol. 26, 563–569. doi: 10.1161/01.ATV.0000203503.82693.c1
Bolós, M., Llorens-Martín, M., Perea, J. R., Jurado-Arjona, J., Rábano, A., Hernández, F., et al. (2017). Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 12:59. doi: 10.1186/s13024-017-0200-1
Borzelleca, J. F., Olson, J. W., and Reno, F. E. (1991). Lifetime toxicity/carcinogenicity studies of FD & C red no. 40 (allura red) in mice. Food Chem. Toxicol. 29, 313–319. doi: 10.1016/0278-6915(91)90202-i
Bradshaw, E. M., Chibnik, L. B., Keenan, B. T., Ottoboni, L., Raj, T., Tang, A., et al. (2013). CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850. doi: 10.1038/nn.3435
Brown, G. C., and Neher, J. J. (2014). Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15, 209–216. doi: 10.1038/nrn3710
Buckingham, J. C., John, C. D., Solito, E., Tierney, T., Flower, R. J., Christian, H., et al. (2006). Annexin 1, glucocorticoids, and the neuroendocrine-immune interface. Ann. N. Y. Acad. Sci. 1088, 396–409. doi: 10.1196/annals.1366.002
Buhl, E., Kim, Y. A., Parsons, T., Zhu, B., Santa-Maria, I., Lefort, R., et al. (2022). Effects of Eph/ephrin signalling and human Alzheimer’s disease-associated EphA1 on Drosophila behaviour and neurophysiology. Neurobiol. Dis. 170:105752. doi: 10.1016/j.nbd.2022.105752
Burk, K., Ramachandran, B., Ahmed, S., Hurtado-Zavala, J. I., Awasthi, A., Benito, E., et al. (2018). Regulation of dendritic spine morphology in hippocampal neurons by Copine-6. Cereb. Cortex 28, 1087–1104. doi: 10.1093/cercor/bhx009
Burnstock, G. (2016). An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 104, 4–17. doi: 10.1016/j.neuropharm.2015.05.031
Burnstock, G., and Knight, G. E. (2018). The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 14, 1–18. doi: 10.1007/s11302-017-9593-0
Burnstock, G., Krügel, U., Abbracchio, M. P., and Illes, P. (2011). Purinergic signalling: from normal behaviour to pathological brain function. Prog. Neurobiol. 95, 229–274. doi: 10.1016/j.pneurobio.2011.08.006
Butler, C. A., Popescu, A. S., Kitchener, E. J. A., Allendorf, D. H., Puigdellívol, M., and Brown, G. C. (2021). Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 158, 621–639. doi: 10.1111/jnc.15327
Campagno, K. E., and Mitchell, C. H. (2021). The P2X(7) receptor in microglial cells modulates the endolysosomal axis, autophagy, and phagocytosis. Front. Cell. Neurosci. 15:645244. doi: 10.3389/fncel.2021.645244
Cantuti-Castelvetri, L., Fitzner, D., Bosch-Queralt, M., Weil, M.-T., Su, M., Sen, P., et al. (2018). Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359, 684–688. doi: 10.1126/science.aan4183
Cao, H., Zhou, X., Chen, Y., Ip, F., Chen, Y., Lai, N. C. H., et al. (2022). Association of SPI1 haplotypes with altered SPI1 gene expression and Alzheimer’s disease risk. J. Alzheimers. Dis. 86, 1861–1873. doi: 10.3233/JAD-215311
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., et al. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924. doi: 10.1038/nn1715
Carta, A. R., and Pisanu, A. (2013). Modulating microglia activity with PPAR-γ agonists: a promising therapy for Parkinson’s disease? Neurotox. Res. 23, 112–123. doi: 10.1007/s12640-012-9342-7
Casali, B. T., MacPherson, K. P., Reed-Geaghan, E. G., and Landreth, G. E. (2020). Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol. Dis. 142:104956. doi: 10.1016/j.nbd.2020.104956
Chan, R. B., Oliveira, T. G., Cortes, E. P., Honig, L. S., Duff, K. E., Small, S. A., et al. (2012). Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 287, 2678–2688. doi: 10.1074/jbc.M111.274142
Chen, B., Huang, S., Su, Y., Wu, Y.-J., Hanna, A., Brickshawana, A., et al. (2019). Macrophage Smad3 protects the infarcted heart, stimulating phagocytosis and regulating inflammation. Circ. Res. 125, 55–70. doi: 10.1161/CIRCRESAHA.119.315069
Chen, K., Zhang, L., Tan, M., Lai, C. S. W., Li, A., Ren, C., et al. (2017). Treadmill exercise suppressed stress-induced dendritic spine elimination in mouse barrel cortex and improved working memory via BDNF/TrkB pathway. Transl. Psychiatry 7:e1069. doi: 10.1038/tp.2017.41
Chen, M., Sinha, M., Luxon, B. A., Bresnick, A. R., and O’Connor, K. L. (2009). Integrin alpha6beta4 controls the expression of genes associated with cell motility, invasion, and metastasis, including S100A4/metastasin. J. Biol. Chem. 284, 1484–1494. doi: 10.1074/jbc.M803997200
Chen, X., Hu, J., Jiang, L., Xu, S., Zheng, B., Wang, C., et al. (2014). Brilliant Blue G improves cognition in an animal model of Alzheimer’s disease and inhibits amyloid-β-induced loss of filopodia and dendrite spines in hippocampal neurons. Neuroscience 279, 94–101. doi: 10.1016/j.neuroscience.2014.08.036
Cheng, Q., Danao, J., Talreja, S., Wen, P., Yin, J., Sun, N., et al. (2018). TREM2-activating antibodies abrogate the negative pleiotropic effects of the Alzheimer’s disease variant Trem2(R47H) on murine myeloid cell function. J. Biol. Chem. 293, 12620–12633. doi: 10.1074/jbc.RA118.001848
Cianciulli, A., Calvello, R., Porro, C., Trotta, T., and Panaro, M. A. (2017). Understanding the role of SOCS signaling in neurodegenerative diseases: current and emerging concepts. Cytokine Growth Factor Rev. 37, 67–79. doi: 10.1016/j.cytogfr.2017.07.005
Cignarella, F., Filipello, F., Bollman, B., Cantoni, C., Locca, A., Mikesell, R., et al. (2020). TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 140, 513–534. doi: 10.1007/s00401-020-02193-z
Claes, C., Danhash, E. P., Hasselmann, J., Chadarevian, J. P., Shabestari, S. K., England, W. E., et al. (2021). Plaque-associated human microglia accumulate lipid droplets in a chimeric model of Alzheimer’s disease. Mol. Neurodegener. 16:50. doi: 10.1186/s13024-021-00473-0
Clayton, K., Enoo, A., and Van Ikezu, T. (2017). Alzheimer’s disease: the role of microglia in brain homeostasis and proteopathy. Front. Neurosci. 11:680. doi: 10.3389/fnins.2017.00680
Cockram, T. O. J., Puigdellívol, M., and Brown, G. C. (2019). Calreticulin and Galectin-3 opsonise bacteria for phagocytosis by microglia. Front. Immunol. 10:2647. doi: 10.3389/fimmu.2019.02647
Colonna, M., and Butovsky, O. (2017). Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
Combs, C. K., Johnson, D. E., Cannady, S. B., Lehman, T. M., and Landreth, G. E. (1999). Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J. Neurosci. 19, 928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., and Small, G. W. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Correale, J. (2014). The role of microglial activation in disease progression. Mult. Scler. 20, 1288–1295. doi: 10.1177/1352458514533230
Cristante, E., McArthur, S., Mauro, C., Maggioli, E., Romero, I. A., Wylezinska-Arridge, M., et al. (2013). Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. Proc. Natl. Acad. Sci. U S A. 110, 832–841. doi: 10.1073/pnas.1209362110
Cui, W., Sun, C., Ma, Y., Wang, S., Wang, X., and Zhang, Y. (2020). Inhibition of TLR4 induces M2 microglial polarization and provides neuroprotection via the NLRP3 inflammasome in Alzheimer’s disease. Front. Neurosci. 14:444. doi: 10.3389/fnins.2020.00444
Curtis, D., Bakaya, K., Sharma, L., and Bandyopadhyay, S. (2020). Weighted burden analysis of exome-sequenced late-onset Alzheimer’s cases and controls provides further evidence for a role for PSEN1 and suggests involvement of the PI3K/Akt/GSK-3β and WNT signalling pathways. Ann. Hum. Genet. 84, 291–302. doi: 10.1111/ahg.12375
Czirr, E., Castello, N. A., Mosher, K. I., Castellano, J. M., Hinkson, I. V., Lucin, K. M., et al. (2017). Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. J. Exp. Med. 214, 1081–1092. doi: 10.1084/jem.20162011
Dagher, N. N., Najafi, A. R., Kayala, K. M. N., Elmore, M. R. P., White, T. E., Medeiros, R., et al. (2015). Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J. Neuroinflamm. 12:139. doi: 10.1186/s12974-015-0366-9
Dalal, P. J., Muller, W. A., and Sullivan, D. P. (2020). Endothelial cell calcium signaling during barrier function and inflammation. Am. J. Pathol. 190, 535–542. doi: 10.1016/j.ajpath.2019.11.004
De Luca, S. N., Soch, A., Sominsky, L., Nguyen, T.-X., Bosakhar, A., and Spencer, S. J. (2020). Glial remodeling enhances short-term memory performance in Wistar rats. J. Neuroinflamm. 17:52. doi: 10.1186/s12974-020-1729-4
Decker, H., Jürgensen, S., Adrover, M. F., Brito-Moreira, J., Bomfim, T. R., Klein, W. L., et al. (2010). N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-β peptide oligomers. J. Neurochem. 115, 1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x
Deczkowska, A., Keren-Shaul, H., Weiner, A., Colonna, M., Schwartz, M., and Amit, I. (2018). Disease-Associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081. doi: 10.1016/j.cell.2018.05.003
Degterev, A., Ofengeim, D., and Yuan, J. (2019). Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. U S A. 116, 9714–9722. doi: 10.1073/pnas.1901179116
Deming, Y., Filipello, F., Cignarella, F., Cantoni, C., Hsu, S., Mikesell, R., et al. (2019). The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 11:eaau2291. doi: 10.1126/scitranslmed.aau2291
Dempsey, C., Rubio Araiz, A., Bryson, K. J., Finucane, O., Larkin, C., Mills, E. L., et al. (2017). Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain. Behav. Immun. 61, 306–316. doi: 10.1016/j.bbi.2016.12.014
Deng, Z., Jia, Y., Liu, H., He, M., Yang, Y., Xiao, W., et al. (2019). RhoA/ROCK pathway: implication in osteoarthritis and therapeutic targets. Am. J. Transl. Res. 11, 5324–5331.
Di Bona, D., Scapagnini, G., Candore, G., Castiglia, L., Colonna-Romano, G., Duro, G., et al. (2010). Immune-inflammatory responses and oxidative stress in Alzheimer’s disease: therapeutic implications. Curr. Pharm. Des. 16, 684–691. doi: 10.2174/138161210790883769
Di Virgilio, F., Dal Ben, D., Sarti, A. C., Giuliani, A. L., and Falzoni, S. (2017). The P2X7 receptor in infection and inflammation. Immunity 47, 15–31. doi: 10.1016/j.immuni.2017.06.020
Di Virgilio, F., Schmalzing, G., and Markwardt, F. (2018). The elusive P2X7 macropore. Trends Cell Biol. 28, 392–404. doi: 10.1016/j.tcb.2018.01.005
Diaz-Aparicio, I., Paris, I., Sierra-Torre, V., Plaza-Zabala, A., Rodríguez-Iglesias, N., Márquez-Ropero, M., et al. (2020). Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J. Neurosci. 40, 1453–1482. doi: 10.1523/JNEUROSCI.0993-19.2019
Dickson, D. W. (1997). The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol. 56, 321–339. doi: 10.1097/00005072-199704000-00001
Domercq, M., and Matute, C. (2019). Targeting P2X4 and P2X7 receptors in multiple sclerosis. Curr. Opin. Pharmacol. 47, 119–125. doi: 10.1016/j.coph.2019.03.010
Du, R.-H., Sun, H.-B., Hu, Z.-L., Lu, M., Ding, J.-H., and Hu, G. (2018). Kir6.1/K-ATP channel modulates microglia phenotypes: implication in Parkinson’s disease. Cell Death Dis. 9:404. doi: 10.1038/s41419-018-0437-9
DuBois, J. C., Ray, A. K., Davies, P., and Shafit-Zagardo, B. (2020). Anti-Axl antibody treatment reduces the severity of experimental autoimmune encephalomyelitis. J. Neuroinflamm. 17:324. doi: 10.1186/s12974-020-01982-3
Durham, G. A., Williams, J. J. L., Nasim, M. T., and Palmer, T. M. (2019). Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol. Sci. 40, 298–308. doi: 10.1016/j.tips.2019.03.001
Efthymiou, A. G., and Goate, A. M. (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12:43. doi: 10.1186/s13024-017-0184-x
Eikelenboom, P., and Veerhuis, R. (1996). The role of complement and activated microglia in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 17, 673–680. doi: 10.1016/0197-4580(96)00108-x
El Khoury, J. B., Moore, K. J., Means, T. K., Leung, J., Terada, K., Toft, M., et al. (2003). CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 197, 1657–1666. doi: 10.1084/jem.20021546
Ellwanger, D. C., Wang, S., Brioschi, S., Shao, Z., Green, L., Case, R., et al. (2021). Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A. 118:e2017742118. doi: 10.1073/pnas.2017742118
Ewers, M., Biechele, G., Suárez-Calvet, M., Sacher, C., Blume, T., Morenas-Rodriguez, E., et al. (2020). Higher CSF sTREM2 and microglia activation are associated with slower rates of beta-amyloid accumulation. EMBO Mol. Med. 12:e12308. doi: 10.15252/emmm.202012308
Ewers, M., Franzmeier, N., Suárez-Calvet, M., Morenas-Rodriguez, E., Caballero, M. A. A., Kleinberger, G., et al. (2019). Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 11:eaav6221. doi: 10.1126/scitranslmed.aav6221
Eysert, F., Coulon, A., Boscher, E., Vreulx, A.-C., Flaig, A., Mendes, T., et al. (2021). Alzheimer’s genetic risk factor FERMT2 (Kindlin-2) controls axonal growth and synaptic plasticity in an APP-dependent manner. Mol. Psychiatry 26, 5592–5607. doi: 10.1038/s41380-020-00926-w
Fabbrizio, P., Amadio, S., Apolloni, S., and Volonté, C. (2017). P2X7 receptor activation modulates autophagy in SOD1-G93A mouse microglia. Front. Cell. Neurosci. 11:249. doi: 10.3389/fncel.2017.00249
Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898. doi: 10.1172/JCI1112
Fan, K.-H., Feingold, E., Rosenthal, S. L., Demirci, F. Y., Ganguli, M., Lopez, O. L., et al. (2020). Whole-Exome sequencing analysis of Alzheimer’s disease in Non-APOE*4 carriers. J. Alzheimers. Dis. 76, 1553–1565. doi: 10.3233/JAD-200037
Fan, Z., Brooks, D. J., Okello, A., and Edison, P. (2017). An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 140, 792–803. doi: 10.1093/brain/aww349
Farfara, D., Trudler, D., Segev-Amzaleg, N., Galron, R., Stein, R., and Frenkel, D. (2011). γ-Secretase component presenilin is important for microglia β-amyloid clearance. Ann. Neurol. 69, 170–180. doi: 10.1002/ana.22191
Feng, J., Wang, J.-X., Du, Y.-H., Liu, Y., Zhang, W., Chen, J.-F., et al. (2018). Dihydromyricetin inhibits microglial activation and neuroinflammation by suppressing NLRP3 inflammasome activation in APP/PS1 transgenic mice. CNS Neurosci. Ther. 24, 1207–1218. doi: 10.1111/cns.12983
Fernandez-Marcos, P. J., and Auwerx, J. (2011). Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, 884S–890S. doi: 10.3945/ajcn.110.001917
Filippini, N., Rao, A., Wetten, S., Gibson, R. A., Borrie, M., Guzman, D., et al. (2009). Anatomically-distinct genetic associations of APOE epsilon4 allele load with regional cortical atrophy in Alzheimer’s disease. Neuroimage 44, 724–728. doi: 10.1016/j.neuroimage.2008.10.003
Fletcher, E. L., Wang, A. Y., Jobling, A. I., Rutar, M. V., Greferath, U., Gu, B., et al. (2019). Targeting P2X7 receptors as a means for treating retinal disease. Drug Discov. Today 24, 1598–1605. doi: 10.1016/j.drudis.2019.03.029
Flores, J., Noël, A., Foveau, B., Lynham, J., Lecrux, C., and LeBlanc, A. C. (2018). Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 9:3916. doi: 10.1038/s41467-018-06449-x
Flowers, S. A., and Rebeck, G. W. (2020). APOE in the normal brain. Neurobiol. Dis. 136:104724. doi: 10.1016/j.nbd.2019.104724
Fonseca, M. I., Ager, R. R., Chu, S.-H., Yazan, O., Sanderson, S. D., LaFerla, F. M., et al. (2009). Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 183, 1375–1383. doi: 10.4049/jimmunol.0901005
Fonseca, M. I., Kawas, C. H., Troncoso, J. C., and Tenner, A. J. (2004a). Neuronal localization of C1q in preclinical Alzheimer’s disease. Neurobiol. Dis. 15, 40–46. doi: 10.1016/j.nbd.2003.09.004
Fonseca, M. I., McGuire, S. O., Counts, S. E., and Tenner, A. J. (2013). Complement activation fragment C5a receptors, CD88 and C5L2, are associated with neurofibrillary pathology. J. Neuroinflamm. 10:25. doi: 10.1186/1742-2094-10-25
Fonseca, M. I., Zhou, J., Botto, M., and Tenner, A. J. (2004b). Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 24, 6457–6465. doi: 10.1523/JNEUROSCI.0901-04.2004
Franklin, R. J. M., and Kotter, M. R. (2008). The biology of CNS remyelination: the key to therapeutic advances. J. Neurol. 255(Suppl.), 19–25. doi: 10.1007/s00415-008-1004-6
Fraser, D. A., and Tenner, A. J. (2008). Directing an appropriate immune response: the role of defense collagens and other soluble pattern recognition molecules. Curr. Drug Targets 9, 113–122. doi: 10.2174/138945008783502476
Fraser, D. A., Pisalyaput, K., and Tenner, A. J. (2010). C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J. Neurochem. 112, 733–743. doi: 10.1111/j.1471-4159.2009.06494.x
Freeman, L., Guo, H., David, C. N., Brickey, W. J., Jha, S., and Ting, J. P.-Y. (2017). NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 214, 1351–1370. doi: 10.1084/jem.20150237
Frenkel, D., Wilkinson, K., Zhao, L., Hickman, S. E., Means, T. K., Puckett, L., et al. (2013). Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 4:2030. doi: 10.1038/ncomms3030
Frey, U., and Morris, R. G. (1998). Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 21, 181–188. doi: 10.1016/s0166-2236(97)01189-2
Friggeri, A., Banerjee, S., Biswas, S., de Freitas, A., Liu, G., Bierhaus, A., et al. (2011). Participation of the receptor for advanced glycation end products in efferocytosis. J. Immunol. 186, 6191–6198. doi: 10.4049/jimmunol.1004134
Fu, A. K. Y., Hung, K.-W., Yuen, M. Y. F., Zhou, X., Mak, D. S. Y., Chan, I. C. W., et al. (2016). IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc. Natl. Acad. Sci. U S A. 113, E2705–E2713. doi: 10.1073/pnas.1604032113
Fu, H., Liu, B., Frost, J. L., Hong, S., Jin, M., Ostaszewski, B., et al. (2012). Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia 60, 993–1003. doi: 10.1002/glia.22331
Fu, Y., Hsiao, J.-H. T., Paxinos, G., Halliday, G. M., and Kim, W. S. (2016). ABCA7 mediates phagocytic clearance of Amyloid-β in the brain. J. Alzheimers. Dis. 54, 569–584. doi: 10.3233/JAD-160456
Gao, P., Ye, L., Cheng, H., and Li, H. (2021). The mechanistic role of bridging integrator 1 (BIN1) in Alzheimer’s disease. Cell. Mol. Neurobiol. 41, 1431–1440. doi: 10.1007/s10571-020-00926-y
Gao, Q., Liang, X., Shaikh, A. S., Zang, J., Xu, W., and Zhang, Y. (2018). JAK/STAT signal transduction: promising attractive targets for immune, inflammatory and hematopoietic diseases. Curr. Drug Targets 19, 487–500. doi: 10.2174/1389450117666161207163054
Gardai, S. J., McPhillips, K. A., Frasch, S. C., Janssen, W. J., Starefeldt, A., Murphy-Ullrich, J. E., et al. (2005). Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334. doi: 10.1016/j.cell.2005.08.032
Ghosh, S., Wu, M. D., Shaftel, S. S., Kyrkanides, S., LaFerla, F. M., Olschowka, J. A., et al. (2013). Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 33, 5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Gipson, C. D., and Olive, M. F. (2017). Structural and functional plasticity of dendritic spines - root or result of behavior? Genes. Brain. Behav. 16, 101–117. doi: 10.1111/gbb.12324
Goldstein, J. L., and Brown, M. S. (2015). A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161, 161–172. doi: 10.1016/j.cell.2015.01.036
Golia, M. T., Poggini, S., Alboni, S., Garofalo, S., Ciano Albanese, N., Viglione, A., et al. (2019). Interplay between inflammation and neural plasticity: both immune activation and suppression impair LTP and BDNF expression. Brain. Behav. Immun. 81, 484–494. doi: 10.1016/j.bbi.2019.07.003
Götzl, J. K., Colombo, A.-V., Fellerer, K., Reifschneider, A., Werner, G., Tahirovic, S., et al. (2018). Early lysosomal maturation deficits in microglia triggers enhanced lysosomal activity in other brain cells of progranulin knockout mice. Mol. Neurodegener. 13:48. doi: 10.1186/s13024-018-0281-5
Griciuc, A., Patel, S., Federico, A. N., Choi, S. H., Innes, B. J., Oram, M. K., et al. (2019). TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron 103, 820–835.e7. doi: 10.1016/j.neuron.2019.06.010.
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Griffin, W. S., Sheng, J. G., Royston, M. C., Gentleman, S. M., McKenzie, J. E., Graham, D. I., et al. (1998). Glial-neuronal interactions in Alzheimer’s disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol. 8, 65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x
Grommes, C., Lee, C. Y. D., Wilkinson, B. L., Jiang, Q., Koenigsknecht-Talboo, J. L., Varnum, B., et al. (2008). Regulation of microglial phagocytosis and inflammatory gene expression by Gas6 acting on the Axl/Mer family of tyrosine kinases. J. neuroimmune Pharmacol. 3, 130–140. doi: 10.1007/s11481-007-9090-2
Gu, B. J., and Wiley, J. S. (2018). P2X7 as a scavenger receptor for innate phagocytosis in the brain. Br. J. Pharmacol. 175, 4195–4208. doi: 10.1111/bph.14470
Gu, B. J., Duce, J. A., Valova, V. A., Wong, B., Bush, A. I., Petrou, S., et al. (2012). P2X7 receptor-mediated scavenger activity of mononuclear phagocytes toward non-opsonized particles and apoptotic cells is inhibited by serum glycoproteins but remains active in cerebrospinal fluid. J. Biol. Chem. 287, 17318–17330. doi: 10.1074/jbc.M112.340885
Gu, B. J., Rathsam, C., Stokes, L., McGeachie, A. B., and Wiley, J. S. (2009). Extracellular ATP dissociates nonmuscle myosin from P2X(7) complex: this dissociation regulates P2X(7) pore formation. Am. J. Physiol. Cell Physiol. 297, C430–C439. doi: 10.1152/ajpcell.00079.2009
Gu, B. J., Saunders, B. M., Jursik, C., and Wiley, J. S. (2010). The P2X7-nonmuscle myosin membrane complex regulates phagocytosis of nonopsonized particles and bacteria by a pathway attenuated by extracellular ATP. Blood 115, 1621–1631. doi: 10.1182/blood-2009-11-251744
Gu, B. J., Saunders, B. M., Petrou, S., and Wiley, J. S. (2011). P2X(7) is a scavenger receptor for apoptotic cells in the absence of its ligand, extracellular ATP. J. Immunol. 187, 2365–2375. doi: 10.4049/jimmunol.1101178
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guo, L., Bertola, D. R., Takanohashi, A., Saito, A., Segawa, Y., Yokota, T., et al. (2019). Bi-allelic CSF1R mutations cause skeletal dysplasia of dysosteosclerosis-pyle disease spectrum and degenerative encephalopathy with brain malformation. Am. J. Hum. Genet. 104, 925–935. doi: 10.1016/j.ajhg.2019.03.004
Guo, S., Wang, H., and Yin, Y. (2022). Microglia polarization from M1 to M2 in neurodegenerative diseases. Front. Aging Neurosci. 14:815347. doi: 10.3389/fnagi.2022.815347
Hammond, J. W., Bellizzi, M. J., Ware, C., Qiu, W. Q., Saminathan, P., Li, H., et al. (2020). Complement-dependent synapse loss and microgliosis in a mouse model of multiple sclerosis. Brain. Behav. Immun. 87, 739–750. doi: 10.1016/j.bbi.2020.03.004
Han, M., Ryu, G., Shin, S.-A., An, J., Kim, H., Park, D., et al. (2021). Physiological roles of apoptotic cell clearance: beyond immune functions. Life (basel) 11:1141. doi: 10.3390/life11111141
Hanayama, R., Tanaka, M., Miwa, K., Shinohara, A., Iwamatsu, A., and Nagata, S. (2002). Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187. doi: 10.1038/417182a
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Hardy, J., and Salih, D. (2021). TREM2-mediated activation of microglia breaks link between amyloid and tau. Lancet. Neurol. 20, 416–417. doi: 10.1016/S1474-4422(21)00133-2
Haslund-Vinding, J., McBean, G., Jaquet, V., and Vilhardt, F. (2017). NADPH oxidases in oxidant production by microglia: activating receptors, pharmacology and association with disease. Br. J. Pharmacol. 174, 1733–1749. doi: 10.1111/bph.13425
He, M., Kubo, H., Morimoto, K., Fujino, N., Suzuki, T., Takahasi, T., et al. (2011). Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 12, 358–364. doi: 10.1038/embor.2011.28
Heckmann, B. L., Boada-Romero, E., Cunha, L. D., Magne, J., and Green, D. R. (2017). LC3-Associated phagocytosis and inflammation. J. Mol. Biol. 429, 3561–3576. doi: 10.1016/j.jmb.2017.08.012
Heneka, M. T., McManus, R. M., and Latz, E. (2018). Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 19, 610–621. doi: 10.1038/s41583-018-0055-7
Heneka, M., Carson, M., El Khoury, J., Landreth, G., Brosseron, F., and Feinstein, D. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405.
Henjum, K., Almdahl, I. S., Årskog, V., Minthon, L., Hansson, O., Fladby, T., et al. (2016). Cerebrospinal fluid soluble TREM2 in aging and Alzheimer’s disease. Alzheimers. Res. Ther. 8:17. doi: 10.1186/s13195-016-0182-1
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Heslegrave, A., Heywood, W., Paterson, R., Magdalinou, N., Svensson, J., Johansson, P., et al. (2016). Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 11:3. doi: 10.1186/s13024-016-0071-x
Hodge, R. D., Bakken, T. E., Miller, J. A., Smith, K. A., Barkan, E. R., Graybuck, L. T., et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. doi: 10.1038/s41586-019-1506-7
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J.-C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43, 429–435. doi: 10.1038/ng.803
Holstege, H. E. (2020). Exome sequencing identifies three novel AD-associated genes. Alzheimer’s Dement. J. Alzheimer’s Assoc. 7, 14–24.
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hoshino, K., Hasegawa, K., Kamiya, H., and Morimoto, Y. (2017). Synapse-specific effects of IL-1β on long-term potentiation in the mouse hippocampus. Biomed. Res. 38, 183–188. doi: 10.2220/biomedres.38.183
Huang, Y., Happonen, K. E., Burrola, P. G., O’Connor, C., Hah, N., Huang, L., et al. (2021). Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 22, 586–594. doi: 10.1038/s41590-021-00913-5
Husemann, J., Loike, J. D., Anankov, R., Febbraio, M., and Silverstein, S. C. (2002). Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia 40, 195–205. doi: 10.1002/glia.10148
Hutter-Paier, B., Huttunen, H. J., Puglielli, L., Eckman, C. B., Kim, D. Y., Hofmeister, A., et al. (2004). The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 44, 227–238. doi: 10.1016/j.neuron.2004.08.043
Huynh, T.-P. V., Davis, A. A., Ulrich, J. D., and Holtzman, D. M. (2017). Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid Res. 58, 824–836. doi: 10.1194/jlr.R075481
Hynes, R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687. doi: 10.1016/s0092-8674(02)00971-6
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Illes, P., Rubini, P., Huang, L., and Tang, Y. (2019a). The P2X7 receptor: a new therapeutic target in Alzheimer’s disease. Expert Opin. Ther. Targets 23, 165–176. doi: 10.1080/14728222.2019.1575811
Illes, P., Verkhratsky, A., and Tang, Y. (2019b). Pathological ATPergic signaling in major depression and bipolar disorder. Front. Mol. Neurosci. 12:331. doi: 10.3389/fnmol.2019.00331
Illes, P., Rubini, P., Ulrich, H., Zhao, Y., and Tang, Y. (2020). Regulation of microglial functions by purinergic mechanisms in the healthy and diseased CNS. Cells 9:1108. doi: 10.3390/cells9051108
Ismail, R., Parbo, P., Madsen, L. S., Hansen, A. K., Hansen, K. V., Schaldemose, J. L., et al. (2020). The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer’s disease: a longitudinal PET study. J. Neuroinflamm. 17:151. doi: 10.1186/s12974-020-01820-6
Jacobi, D., Stanya, K. J., and Lee, C.-H. (2012). Adipose tissue signaling by nuclear receptors in metabolic complications of obesity. Adipocyte 1, 4–12. doi: 10.4161/adip.19036
Jehle, A. W., Gardai, S. J., Li, S., Linsel-Nitschke, P., Morimoto, K., Janssen, W. J., et al. (2006). ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 174, 547–556. doi: 10.1083/jcb.200601030
Ji, J., Xue, T.-F., Guo, X.-D., Yang, J., Guo, R.-B., Wang, J., et al. (2018). Antagonizing peroxisome proliferator-activated receptor γ facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell 17:e12774. doi: 10.1111/acel.12774
Jiang, C.-T., Wu, W.-F., Deng, Y.-H., and Ge, J.-W. (2020). Modulators of microglia activation and polarization in ischemic stroke (Review). Mol. Med. Rep. 21, 2006–2018. doi: 10.3892/mmr.2020.11003
Jiang, H., Burdick, D., Glabe, C. G., Cotman, C. W., and Tenner, A. J. (1994). beta-Amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J. Immunol. 152, 5050–5059.
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Jung, J.-S., Choi, M.-J., Lee, Y. Y., Moon, B.-I., Park, J.-S., and Kim, H.-S. (2017). Suppression of lipopolysaccharide-induced neuroinflammation by morin via MAPK, PI3K/Akt, and PKA/HO-1 signaling pathway modulation. J. Agric. Food Chem. 65, 373–382. doi: 10.1021/acs.jafc.6b05147
Kajiho, H., Sakurai, K., Minoda, T., Yoshikawa, M., Nakagawa, S., Fukushima, S., et al. (2011). Characterization of RIN3 as a guanine nucleotide exchange factor for the Rab5 subfamily GTPase Rab31. J. Biol. Chem. 286, 24364–24373. doi: 10.1074/jbc.M110.172445
Kaminski, W. E., Orsó, E., Diederich, W., Klucken, J., Drobnik, W., and Schmitz, G. (2000). Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7). Biochem. Biophys. Res. Commun. 273, 532–538. doi: 10.1006/bbrc.2000.2954
Kanekiyo, T., and Bu, G. (2014). The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 6:93. doi: 10.3389/fnagi.2014.00093
Karch, C. M., Ezerskiy, L. A., Bertelsen, S., and Goate, A. M. (2016). Alzheimer’s disease risk polymorphisms regulate gene expression in the ZCWPW1 and the CELF1 loci. PLoS One 11:e0148717. doi: 10.1371/journal.pone.0148717
Karch, C. M., Jeng, A. T., Nowotny, P., Cady, J., Cruchaga, C., and Goate, A. M. (2012). Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS One 7:e50976. doi: 10.1371/journal.pone.0050976
Kaushal, V., Dye, R., Pakavathkumar, P., Foveau, B., Flores, J., Hyman, B., et al. (2015). Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 22, 1676–1686. doi: 10.1038/cdd.2015.16
Kawabe, K., Takano, K., Moriyama, M., and Nakamura, Y. (2018). Microglia endocytose Amyloid β through the binding of transglutaminase 2 and milk fat globule EGF factor 8 protein. Neurochem. Res. 43, 41–49. doi: 10.1007/s11064-017-2284-y
Kawai, T., and Akira, S. (2007). Signaling to NF-kappaB by toll-like receptors. Trends Mol. Med. 13, 460–469. doi: 10.1016/j.molmed.2007.09.002
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
Kim, A., García-García, E., Straccia, M., Comella-Bolla, A., Miguez, A., Masana, M., et al. (2020). Reduced fractalkine levels lead to striatal synaptic plasticity deficits in Huntington’s disease. Front. Cell. Neurosci. 14:163. doi: 10.3389/fncel.2020.00163
Kleinberger, G., Brendel, M., Mracsko, E., Wefers, B., Groeneweg, L., Xiang, X., et al. (2017). The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 36, 1837–1853. doi: 10.15252/embj.201796516
Kober, D. L., Alexander-Brett, J. M., Karch, C. M., Cruchaga, C., Colonna, M., Holtzman, M. J., et al. (2016). Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife 5:e20391. doi: 10.7554/eLife.20391
Koenigsknecht, J., and Landreth, G. (2004). Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J. Neurosci. 24, 9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004
Kojima, Y., Weissman, I. L., and Leeper, N. J. (2017). The role of efferocytosis in atherosclerosis. Circulation 135, 476–489.
Koldamova, R. P., Lefterov, I. M., Ikonomovic, M. D., Skoko, J., Lefterov, P. I., Isanski, B. A., et al. (2003). 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J. Biol. Chem. 278, 13244–13256. doi: 10.1074/jbc.M300044200
Korb, L. C., and Ahearn, J. M. (1997). C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J. Immunol. 158, 4525–4528.
Koronyo, Y., Salumbides, B. C., Sheyn, J., Pelissier, L., Li, S., Ljubimov, V., et al. (2015). Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain 138, 2399–2422. doi: 10.1093/brain/awv150
Koutsodendris, N., Nelson, M. R., Rao, A., and Huang, Y. (2022). Apolipoprotein E and Alzheimer’s disease: findings, hypotheses, and potential mechanisms. Annu. Rev. Pathol. 17, 73–99. doi: 10.1146/annurev-pathmechdis-030421-112756
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9. doi: 10.1016/j.immuni.2017.08.008.
Kuhn, P.-H., Colombo, A. V., Schusser, B., Dreymueller, D., Wetzel, S., Schepers, U., et al. (2016). Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife 5:e12748. doi: 10.7554/eLife.12748
Kulkarni, B., Cruz-Martins, N., and Kumar, D. (2022). Microglia in Alzheimer’s disease: an unprecedented opportunity as prospective drug target. Mol. Neurobiol. 59, 2678–2693. doi: 10.1007/s12035-021-02661-x
La Rosa, F., Saresella, M., Marventano, I., Piancone, F., Ripamonti, E., Al-Daghri, N., et al. (2019). Stavudine reduces NLRP3 inflammasome activation and modulates Amyloid-β autophagy. J. Alzheimers. Dis. 72, 401–412. doi: 10.3233/JAD-181259
Lacor, P. N., Buniel, M. C., Chang, L., Fernandez, S. J., Gong, Y., Viola, K. L., et al. (2004). Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 24, 10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lan, X., Han, X., Li, Q., Yang, Q.-W., and Wang, J. (2017). Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 13, 420–433. doi: 10.1038/nrneurol.2017.69
Lane, D. J. R., Ayton, S., and Bush, A. I. (2018). Iron and Alzheimer’s disease: an update on emerging mechanisms. J. Alzheimers. Dis. 64, S379–S395. doi: 10.3233/JAD-179944
Lazdon, E., Stolero, N., and Frenkel, D. (2020). Microglia and Parkinson’s disease: footprints to pathology. J. Neural Transm. 127, 149–158. doi: 10.1007/s00702-020-02154-6
Le, Y., Gong, W., Tiffany, H. L., Tumanov, A., Nedospasov, S., Shen, W., et al. (2001). Amyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J. Neurosci. 21:RC123. doi: 10.1523/JNEUROSCI.21-02-j0003.2001
Leduc, V., Jasmin-Bélanger, S., and Poirier, J. (2010). APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med. 16, 469–477. doi: 10.1016/j.molmed.2010.07.008
Lee, C. Y. D., Daggett, A., Gu, X., Jiang, L.-L., Langfelder, P., Li, X., et al. (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97, 1032–1048.e5. doi: 10.1016/j.neuron.2018.02.002.
Lee, C.-M., Lee, D.-S., Jung, W.-K., Yoo, J. S., Yim, M.-J., Choi, Y. H., et al. (2016). Benzyl isothiocyanate inhibits inflammasome activation in E. coli LPS-stimulated BV2 cells. Int. J. Mol. Med. 38, 912–918. doi: 10.3892/ijmm.2016.2667
Leng, F., and Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi: 10.1038/s41582-020-00435-y
Lepeta, K., Lourenco, M. V., Schweitzer, B. C., Martino Adami, P. V., Banerjee, P., Catuara-Solarz, S., et al. (2016). Synaptopathies: synaptic dysfunction in neurological disorders - a review from students to students. J. Neurochem. 138, 785–805. doi: 10.1111/jnc.13713
Lew, D. S., Mazzoni, F., and Finnemann, S. C. (2020). Microglia inhibition delays retinal degeneration due to MerTK phagocytosis receptor deficiency. Front. Immunol. 11:1463. doi: 10.3389/fimmu.2020.01463
Li, E., Noda, M., Doi, Y., Parajuli, B., Kawanokuchi, J., Sonobe, Y., et al. (2012). The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid β toxicity. J. Neuroinflamm. 9:148. doi: 10.1186/1742-2094-9-148
Li, H., Xue, X., Li, L., Li, Y., and Wang Yanni, et al. (2020). Aluminum-Induced synaptic plasticity impairment via PI3K-Akt-mTOR signaling pathway. Neurotox. Res. 37, 996–1008. doi: 10.1007/s12640-020-00165-5
Li, J.-J., Liu, S.-J., Liu, X.-Y., and Ling, E.-A. (2018). Herbal compounds with special reference to gastrodin as potential therapeutic agents for microglia mediated neuroinflammation. Curr. Med. Chem. 25, 5958–5974. doi: 10.2174/0929867325666180214123929
Li, M., Toombes, G. E. S., Silberberg, S. D., and Swartz, K. J. (2015). Physical basis of apparent pore dilation of ATP-activated P2X receptor channels. Nat. Neurosci. 18, 1577–1583. doi: 10.1038/nn.4120
Li, Q., Chen, L., Liu, X., Li, X., Cao, Y., Bai, Y., et al. (2018). Pterostilbene inhibits amyloid-β-induced neuroinflammation in a microglia cell line by inactivating the NLRP3/caspase-1 inflammasome pathway. J. Cell. Biochem. 119, 7053–7062. doi: 10.1002/jcb.27023
Li, S., Jin, M., Koeglsperger, T., Shepardson, N. E., Shankar, G. M., and Selkoe, D. J. (2011). Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 31, 6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011
Li, Y.-Q., Tan, M.-S., Wang, H.-F., Tan, C.-C., Zhang, W., Zheng, Z.-J., et al. (2016). Common variant in PTK2B is associated with late-onset Alzheimer’s disease: a replication study and meta-analyses. Neurosci. Lett. 621, 83–87. doi: 10.1016/j.neulet.2016.04.020
Liao, Y.-L., Zhou, X.-Y., Ji, M.-H., Qiu, L.-C., Chen, X.-H., Gong, C.-S., et al. (2021). S100A9 upregulation contributes to learning and memory impairments by promoting microglia M1 polarization in sepsis survivor mice. Inflammation 44, 307–320. doi: 10.1007/s10753-020-01334-6
Lim, T. K., and Ruthazer, E. S. (2021). Microglial trogocytosis and the complement system regulate axonal pruning in vivo. Elife 10:e62167. doi: 10.7554/eLife.62167
Lin, L., Chen, X., Zhou, Q., Huang, P., Jiang, S., Wang, H., et al. (2019). Synaptic structure and alterations in the hippocampus in neonatal rats exposed to lipopolysaccharide. Neurosci. Lett. 709:134364. doi: 10.1016/j.neulet.2019.134364
Lin, P. B.-C., Tsai, A. P.-Y., Soni, D., Lee-Gosselin, A., Moutinho, M., Puntambekar, S. S., et al. (2022). INPP5D deficiency attenuates amyloid pathology in a mouse model of Alzheimer’s disease. Alzheimers. Dement. Online ahead of print. doi: 10.1002/alz.12849
Lin, Y.-T., Seo, J., Gao, F., Feldman, H. M., Wen, H.-L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-Derived brain cell types. Neuron 98, 1141–1154.e7. doi: 10.1016/j.neuron.2018.05.008.
Linnartz, B., and Neumann, H. (2013). Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia 61, 37–46. doi: 10.1002/glia.22359
Linton, M. F., Moslehi, J. J., and Babaev, V. R. (2019). Akt signaling in macrophage polarization, survival, and atherosclerosis. Int. J. Mol. Sci. 20:2703. doi: 10.3390/ijms20112703
Lisman, J., Cooper, K., Sehgal, M., and Silva, A. J. (2018). Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat. Neurosci. 21, 309–314. doi: 10.1038/s41593-018-0076-6
Liu, W., Taso, O., Wang, R., Bayram, S., Graham, A. C., Garcia-Reitboeck, P., et al. (2020). Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Hum. Mol. Genet. 29, 3224–3248. doi: 10.1093/hmg/ddaa209
Liu, Y.-J., and Chern, Y. (2015). AMPK-mediated regulation of neuronal metabolism and function in brain diseases. J. Neurogenet. 29, 50–58. doi: 10.3109/01677063.2015.1067203
Lucin, K. M., O’Brien, C. E., Bieri, G., Czirr, E., Mosher, K. I., Abbey, R. J., et al. (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79, 873–886. doi: 10.1016/j.neuron.2013.06.046
Lynch, J., Guo, L., Gelebart, P., Chilibeck, K., Xu, J., Molkentin, J. D., et al. (2005). Calreticulin signals upstream of calcineurin and MEF2C in a critical Ca(2+)-dependent signaling cascade. J. Cell Biol. 170, 37–47. doi: 10.1083/jcb.200412156
Ma, C., Li, S., Hu, Y., Ma, Y., Wu, Y., Wu, C., et al. (2021). AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J. Exp. Med. 218:e20201796. doi: 10.1084/jem.20201796
Ma, K., Guo, J., Wang, G., Ni, Q., and Liu, X. (2020). Toll-Like receptor 2-Mediated autophagy promotes microglial cell death by modulating the microglial M1/M2 phenotype. Inflammation 43, 701–711. doi: 10.1007/s10753-019-01152-5
Ma, W., Rai, V., Hudson, B. I., Song, F., Schmidt, A. M., and Barile, G. R. (2012). RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol. 274, 72–82. doi: 10.1016/j.cellimm.2012.02.001
Maccioni, R. B., González, A., Andrade, V., Cortés, N., Tapia, J. P., and Guzmán-Martínez, L. (2018). Alzheimer’s disease in the perspective of neuroimmunology. Open Neurol. J. 12, 50–56. doi: 10.2174/1874205X01812010050
Madry, C., Kyrargyri, V., Arancibia-Cárcamo, I. L., Jolivet, R., Kohsaka, S., Bryan, R. M., et al. (2018). Microglial ramification, surveillance, and Interleukin-1β release are regulated by the two-pore domain K(+) channel THIK-1. Neuron 97, 299–312.e6. doi: 10.1016/j.neuron.2017.12.002.
Maier, M., Peng, Y., Jiang, L., Seabrook, T. J., Carroll, M. C., and Lemere, C. A. (2008). Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J. Neurosci. 28, 6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008
Majumdar, A., Capetillo-Zarate, E., Cruz, D., Gouras, G. K., and Maxfield, F. R. (2011). Degradation of Alzheimer’s amyloid fibrils by microglia requires delivery of ClC-7 to lysosomes. Mol. Biol. Cell 22, 1664–1676. doi: 10.1091/mbc.E10-09-0745
Majumder, S., Crabtree, J. S., Golde, T. E., Minter, L. M., Osborne, B. A., and Miele, L. (2021). Targeting Notch in oncology: the path forward. Nat. Rev. Drug Discov. 20, 125–144. doi: 10.1038/s41573-020-00091-3
Malik, M., Simpson, J. F., Parikh, I., Wilfred, B. R., Fardo, D. W., Nelson, P. T., et al. (2013). CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci. 33, 13320–13325. doi: 10.1523/JNEUROSCI.1224-13.2013
Malireddi, R. K. S., Gurung, P., Kesavardhana, S., Samir, P., Burton, A., Mummareddy, H., et al. (2020). Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 217:jem.20191644. doi: 10.1084/jem.20191644
Malm, T., Koistinaho, M., Muona, A., Magga, J., and Koistinaho, J. (2010). The role and therapeutic potential of monocytic cells in Alzheimer’s disease. Glia 58, 889–900. doi: 10.1002/glia.20973
Manning, B. D., and Toker, A. (2017). AKT/PKB signaling: navigating the network. Cell 169, 381–405. doi: 10.1016/j.cell.2017.04.001
Marcelo, K. L., Means, A. R., and York, B. (2016). The Ca(2+)/Calmodulin/CaMKK2 axis: nature’s metabolic CaMshaft. Trends Endocrinol. Metab. 27, 706–718. doi: 10.1016/j.tem.2016.06.001
Martinez, J., Cunha, L. D., Park, S., Yang, M., Lu, Q., Orchard, R., et al. (2016). Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533, 115–119. doi: 10.1038/nature17950
Masuda, T., Sankowski, R., Staszewski, O., and Prinz, M. (2020). Microglia heterogeneity in the single-cell era. Cell Rep. 30, 1271–1281. doi: 10.1016/j.celrep.2020.01.010
Mathys, H., Adaikkan, C., Gao, F., Young, J. Z., Manet, E., Hemberg, M., et al. (2017). Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 21, 366–380. doi: 10.1016/j.celrep.2017.09.039
Matlung, H. L., Babes, L., Zhao, X. W., van Houdt, M., Treffers, L. W., van Rees, D. J., et al. (2018). Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep. 23, 3946–3959.e6. doi: 10.1016/j.celrep.2018.05.082.
Maxeiner, H., Husemann, J., Thomas, C. A., Loike, J. D., El Khoury, J., and Silverstein, S. C. (1998). Complementary roles for scavenger receptor A and CD36 of human monocyte-derived macrophages in adhesion to surfaces coated with oxidized low-density lipoproteins and in secretion of H2O2. J. Exp. Med. 188, 2257–2265. doi: 10.1084/jem.188.12.2257
Mazaheri, F., Snaidero, N., Kleinberger, G., Madore, C., Daria, A., Werner, G., et al. (2017). TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 18, 1186–1198. doi: 10.15252/embr.201743922
McArthur, S., Cristante, E., Paterno, M., Christian, H., Roncaroli, F., Gillies, G. E., et al. (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol. 185, 6317–6328. doi: 10.4049/jimmunol.1001095
McDade, E., Llibre-Guerra, J. J., Holtzman, D. M., Morris, J. C., and Bateman, R. J. (2021). The informed road map to prevention of Alzheimer disease: a call to arms. Mol. Neurodegener. 16:49. doi: 10.1186/s13024-021-00467-y
McQuade, A., and Blurton-Jones, M. (2019). Microglia in Alzheimer’s disease: exploring how genetics and phenotype influence risk. J. Mol. Biol. 431, 1805–1817. doi: 10.1016/j.jmb.2019.01.045
Medina, M., and Avila, J. (2014). The role of extracellular Tau in the spreading of neurofibrillary pathology. Front. Cell. Neurosci. 8:113. doi: 10.3389/fncel.2014.00113
Meilandt, W. J., Ngu, H., Gogineni, A., Lalehzadeh, G., Lee, S.-H., Srinivasan, K., et al. (2020). Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J. Neurosci. 40, 1956–1974. doi: 10.1523/JNEUROSCI.1871-19.2019
Mishra, S., Blazey, T. M., Holtzman, D. M., Cruchaga, C., Su, Y., Morris, J. C., et al. (2018). Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE ε4 genotype. Brain 141, 1828–1839. doi: 10.1093/brain/awy103
Mishra, S., Knupp, A., Szabo, M. P., Williams, C. A., Kinoshita, C., Hailey, D. W., et al. (2022). The Alzheimer’s gene SORL1 is a regulator of endosomal traffic and recycling in human neurons. Cell. Mol. Life Sci. 79:162. doi: 10.1007/s00018-022-04182-9
Miyanishi, M., Tada, K., Koike, M., Uchiyama, Y., Kitamura, T., and Nagata, S. (2007). Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439. doi: 10.1038/nature06307
Mohammadi, M., Manaheji, H., Maghsoudi, N., Danyali, S., Baniasadi, M., and Zaringhalam, J. (2020). Microglia dependent BDNF and proBDNF can impair spatial memory performance during persistent inflammatory pain. Behav. Brain Res. 390:112683. doi: 10.1016/j.bbr.2020.112683
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. doi: 10.1016/j.cell.2011.04.005
Morrissey, M. A., Williamson, A. P., Steinbach, A. M., Roberts, E. W., Kern, N., Headley, M. B., et al. (2018). Chimeric antigen receptors that trigger phagocytosis. Elife 7:e36688. doi: 10.7554/eLife.36688
Moutinho, M., and Landreth, G. E. (2017). Therapeutic potential of nuclear receptor agonists in Alzheimer’s disease. J. Lipid Res. 58, 1937–1949. doi: 10.1194/jlr.R075556
Mulder, S. D., Nielsen, H. M., Blankenstein, M. A., Eikelenboom, P., and Veerhuis, R. (2014). Apolipoproteins E and J interfere with amyloid-beta uptake by primary human astrocytes and microglia in vitro. Glia 62, 493–503. doi: 10.1002/glia.22619
Mulero, M. C., Huxford, T., and Ghosh, G. (2019). NF-κB, IκB, and IKK: integral components of immune system signaling. Adv. Exp. Med. Biol. 1172, 207–226. doi: 10.1007/978-981-13-9367-9_10
Muñoz-Planillo, R., Kuffa, P., Martínez-Colón, G., Smith, B. L., Rajendiran, T. M., and Núñez, G. (2013). K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. doi: 10.1016/j.immuni.2013.05.016
Naito, A. T., Sumida, T., Nomura, S., Liu, M.-L., Higo, T., Nakagawa, A., et al. (2012). Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell 149, 1298–1313. doi: 10.1016/j.cell.2012.03.047
Neher, J. J., Neniskyte, U., Zhao, J.-W., Bal-Price, A., Tolkovsky, A. M., and Brown, G. C. (2011). Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 186, 4973–4983. doi: 10.4049/jimmunol.1003600
Nemetchek, M. D., Stierle, A. A., Stierle, D. B., and Lurie, D. I. (2017). The Ayurvedic plant Bacopa monnieri inhibits inflammatory pathways in the brain. J. Ethnopharmacol. 197, 92–100. doi: 10.1016/j.jep.2016.07.073
Nguyen, P. T., Dorman, L. C., Pan, S., Vainchtein, I. D., Han, R. T., Nakao-Inoue, H., et al. (2020). Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 182, 388–403.e15. doi: 10.1016/j.cell.2020.05.050.
Nomoto, M., and Inokuchi, K. (2018). Behavioral, cellular, and synaptic tagging frameworks. Neurobiol. Learn. Mem. 153, 13–20. doi: 10.1016/j.nlm.2018.03.010
Nomura, S., Tandon, N. N., Nakamura, T., Cone, J., Fukuhara, S., and Kambayashi, J. (2001). High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis 158, 277–287. doi: 10.1016/s0021-9150(01)00433-6
Nowell, C. S., and Radtke, F. (2017). Notch as a tumour suppressor. Nat. Rev. Cancer 17, 145–159. doi: 10.1038/nrc.2016.145
Nugent, A. A., Lin, K., van Lengerich, B., Lianoglou, S., Przybyla, L., Davis, S. S., et al. (2020). TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854.e9. doi: 10.1016/j.neuron.2019.12.007.
Okuda, K., Højgaard, K., Privitera, L., Bayraktar, G., and Takeuchi, T. (2021). Initial memory consolidation and the synaptic tagging and capture hypothesis. Eur. J. Neurosci. 54, 6826–6849. doi: 10.1111/ejn.14902
Olmos-Alonso, A., Schetters, S. T. T., Sri, S., Askew, K., Mancuso, R., Vargas-Caballero, M., et al. (2016). Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 139, 891–907. doi: 10.1093/brain/awv379
Olzmann, J. A., and Carvalho, P. (2019). Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 20, 137–155. doi: 10.1038/s41580-018-0085-z
Oosterhof, N., Chang, I. J., Karimiani, E. G., Kuil, L. E., Jensen, D. M., Daza, R., et al. (2019). Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am. J. Hum. Genet. 104, 936–947. doi: 10.1016/j.ajhg.2019.03.010
Origlia, N., Bonadonna, C., Rosellini, A., Leznik, E., Arancio, O., Yan, S. S., et al. (2010). Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J. Neurosci. 30, 11414–11425. doi: 10.1523/JNEUROSCI.2127-10.2010
Orre, M., Kamphuis, W., Osborn, L. M., Jansen, A. H. P., Kooijman, L., Bossers, K., et al. (2014). Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760. doi: 10.1016/j.neurobiolaging.2014.06.004
Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., et al. (2002). Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662. doi: 10.1086/342259
Pan, X., Zhu, Y., Lin, N., Zhang, J., Ye, Q. Y., Huang, H. P., et al. (2011). Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer’s disease. Mol. Neurodegener. 6:45. doi: 10.1186/1750-1326-6-45
Pan, Y., and Monje, M. (2020). Activity shapes neural circuit form and function: a historical perspective. J. Neurosci. 40, 944–954. doi: 10.1523/JNEUROSCI.0740-19.2019
Pang, Q.-M., Chen, S.-Y., Xu, Q.-J., Zhang, M., Liang, D.-F., Fu, S.-P., et al. (2022). Effects of astrocytes and microglia on neuroinflammation after spinal cord injury and related immunomodulatory strategies. Int. Immunopharmacol. 108:108754. doi: 10.1016/j.intimp.2022.108754
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Paolicelli, R. C., Jawaid, A., Henstridge, C. M., Valeri, A., Merlini, M., Robinson, J. L., et al. (2017). TDP-43 depletion in microglia promotes amyloid clearance but also induces synapse loss. Neuron 95, 297–308.e6. doi: 10.1016/j.neuron.2017.05.037.
Paouri, E., Tzara, O., Kartalou, G.-I., Zenelak, S., and Georgopoulos, S. (2017a). Peripheral tumor necrosis factor-alpha (TNF-α) modulates amyloid pathology by regulating blood-derived immune cells and glial response in the brain of AD/TNF transgenic mice. J. Neurosci. 37, 5155–5171. doi: 10.1523/JNEUROSCI.2484-16.2017
Paouri, E., Tzara, O., Zenelak, S., and Georgopoulos, S. (2017b). Genetic deletion of tumor necrosis Factor-α attenuates Amyloid-β production and decreases amyloid plaque formation and glial response in the 5XFAD model of Alzheimer’s disease. J. Alzheimers. Dis. 60, 165–181. doi: 10.3233/JAD-170065
Parhizkar, S., Arzberger, T., Brendel, M., Kleinberger, G., Deussing, M., Focke, C., et al. (2019). Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 22, 191–204. doi: 10.1038/s41593-018-0296-9
Pascoal, T. A., Benedet, A. L., Ashton, N. J., Kang, M. S., Therriault, J., Chamoun, M., et al. (2021). Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 27, 1592–1599. doi: 10.1038/s41591-021-01456-w
Peng, J., Liu, Y., Umpierre, A. D., Xie, M., Tian, D.-S., Richardson, J. R., et al. (2019). Microglial P2Y12 receptor regulates ventral hippocampal CA1 neuronal excitability and innate fear in mice. Mol. Brain 12:71. doi: 10.1186/s13041-019-0492-x
Perregaux, D. G., and Gabel, C. A. (1998). Post-translational processing of murine IL-1: evidence that ATP-induced release of IL-1 alpha and IL-1 beta occurs via a similar mechanism. J. Immunol. 160, 2469–2477.
Perretti, M., and D’Acquisto, F. (2009). Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 9, 62–70. doi: 10.1038/nri2470
Perretti, M., Ingegnoli, F., Wheller, S. K., Blades, M. C., Solito, E., and Pitzalis, C. (2002). Annexin 1 modulates monocyte-endothelial cell interaction in vitro and cell migration in vivo in the human SCID mouse transplantation model. J. Immunol. 169, 2085–2092. doi: 10.4049/jimmunol.169.4.2085
Peters, D. G., Pollack, A. N., Cheng, K. C., Sun, D., Saido, T., Haaf, M. P., et al. (2018). Dietary lipophilic iron alters amyloidogenesis and microglial morphology in Alzheimer’s disease knock-in APP mice. Metallomics 10, 426–443. doi: 10.1039/c8mt00004b
Peterson, T. S., Camden, J. M., Wang, Y., Seye, C. I., Wood, W. G., Sun, G. Y., et al. (2010). P2Y2 nucleotide receptor-mediated responses in brain cells. Mol. Neurobiol. 41, 356–366. doi: 10.1007/s12035-010-8115-7
Pettigrew, L. C., Kryscio, R. J., and Norris, C. M. (2016). The TNFα-Transgenic rat: hippocampal synaptic integrity, cognition, function, and post-ischemic cell loss. PLoS One 11:e0154721. doi: 10.1371/journal.pone.0154721
Piccio, L., Deming, Y., Del-Águila, J. L., Ghezzi, L., Holtzman, D. M., Fagan, A. M., et al. (2016). Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 131, 925–933. doi: 10.1007/s00401-016-1533-5
Pisanu, A., Lecca, D., Mulas, G., Wardas, J., Simbula, G., Spiga, S., et al. (2014). Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis. 71, 280–291. doi: 10.1016/j.nbd.2014.08.011
Plemel, J. R., Michaels, N. J., Weishaupt, N., Caprariello, A. V., Keough, M. B., Rogers, J. A., et al. (2018). Mechanisms of lysophosphatidylcholine-induced demyelination: a primary lipid disrupting myelinopathy. Glia 66, 327–347. doi: 10.1002/glia.23245
Poliani, P., Wang, Y., Fontana, E., Robinette, M., Yamanishi, Y., Gilfillan, S., et al. (2015). TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 125, 2161–2170. doi: 10.1172/JCI77983
Polvikoski, T., Sulkava, R., Haltia, M., Kainulainen, K., Vuorio, A., Verkkoniemi, A., et al. (1995). Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N. Engl. J. Med. 333, 1242–1247. doi: 10.1056/NEJM199511093331902
Poo, M.-M., Pignatelli, M., Ryan, T. J., Tonegawa, S., Bonhoeffer, T., Martin, K. C., et al. (2016). What is memory? the present state of the engram. BMC Biol. 14:40. doi: 10.1186/s12915-016-0261-6
Premkumar, D. R., Cohen, D. L., Hedera, P., Friedland, R. P., and Kalaria, R. N. (1996). Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am. J. Pathol. 148, 2083–2095.
Price, B. R., Sudduth, T. L., Weekman, E. M., Johnson, S., Hawthorne, D., Woolums, A., et al. (2020). Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflamm. 17:238. doi: 10.1186/s12974-020-01915-0
Prokopenko, D., Morgan, S. L., Mullin, K., Hofmann, O., Chapman, B., Kirchner, R., et al. (2021). Whole-genome sequencing reveals new Alzheimer’s disease-associated rare variants in loci related to synaptic function and neuronal development. Alzheimers. Dement. 17, 1509–1527. doi: 10.1002/alz.12319
Puertollano, R., Ferguson, S. M., Brugarolas, J., and Ballabio, A. (2018). The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 37:e98804. doi: 10.15252/embj.201798804
Puglielli, L., Konopka, G., Pack-Chung, E., Ingano, L. A., Berezovska, O., Hyman, B. T., et al. (2001). Acyl-coenzyme a: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 3, 905–912. doi: 10.1038/ncb1001-905
Raghavan, N. S., Brickman, A. M., Andrews, H., Manly, J. J., Schupf, N., Lantigua, R., et al. (2018). Whole-exome sequencing in 20,197 persons for rare variants in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 5, 832–842. doi: 10.1002/acn3.582
Raghuraman, R., Karthikeyan, A., Wei, W. L., Dheen, S. T., and Sajikumar, S. (2019). Activation of microglia in acute hippocampal slices affects activity-dependent long-term potentiation and synaptic tagging and capture in area CA1. Neurobiol. Learn. Mem. 163:107039. doi: 10.1016/j.nlm.2019.107039
Rajbhandari, L., Tegenge, M. A., Shrestha, S., Ganesh Kumar, N., Malik, A., Mithal, A., et al. (2014). Toll-like receptor 4 deficiency impairs microglial phagocytosis of degenerating axons. Glia 62, 1982–1991. doi: 10.1002/glia.22719
Ramsborg, C. G., and Papoutsakis, E. T. (2007). Global transcriptional analysis delineates the differential inflammatory response interleukin-15 elicits from cultured human T cells. Exp. Hematol. 35, 454–464. doi: 10.1016/j.exphem.2006.11.013
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Rauti, R., Cellot, G., D’Andrea, P., Colliva, A., Scaini, D., Tongiorgi, E., et al. (2020). BDNF impact on synaptic dynamics: extra or intracellular long-term release differently regulates cultured hippocampal synapses. Mol. Brain 13:43. doi: 10.1186/s13041-020-00582-9
Reed-Geaghan, E. G., Savage, J. C., Hise, A. G., and Landreth, G. E. (2009). CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 29, 11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009
Reiman, E. M., Chen, K., Liu, X., Bandy, D., Yu, M., Lee, W., et al. (2009). Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A. 106, 6820–6825. doi: 10.1073/pnas.0900345106
Ries, M., and Sastre, M. (2016). Mechanisms of abeta clearance and degradation by glial cells. Front. Aging Neurosci. 8:160. doi: 10.3389/fnagi.2016.00160
Rosin, J. M., Marsters, C. M., Malik, F., Far, R., Adnani, L., Schuurmans, C., et al. (2021). Embryonic microglia interact with hypothalamic radial glia during development and upregulate the TAM receptors MERTK and AXL following an insult. Cell Rep. 34:108587. doi: 10.1016/j.celrep.2020.108587
Ruganzu, J. B., Zheng, Q., Wu, X., He, Y., Peng, X., Jin, H., et al. (2021). TREM2 overexpression rescues cognitive deficits in APP/PS1 transgenic mice by reducing neuroinflammation via the JAK/STAT/SOCS signaling pathway. Exp. Neurol. 336:113506. doi: 10.1016/j.expneurol.2020.113506
Ryu, J. K., and McLarnon, J. G. (2008). Block of purinergic P2X(7) receptor is neuroprotective in an animal model of Alzheimer’s disease. Neuroreport 19, 1715–1719. doi: 10.1097/WNR.0b013e3283179333
Safaiyan, S., Kannaiyan, N., Snaidero, N., Brioschi, S., Biber, K., Yona, S., et al. (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998. doi: 10.1038/nn.4325
Safieh, M., Korczyn, A. D., and Michaelson, D. M. (2019). ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med. 17:64. doi: 10.1186/s12916-019-1299-4
Saito, T., and Saido, T. C. (2018). Neuroinflammation in mouse models of Alzheimer’s disease. Clin. Exp. Neuroimmunol. 9, 211–218. doi: 10.1111/cen3.12475
Salih, D. A., Bayram, S., Guelfi, S., Reynolds, R. H., Shoai, M., Ryten, M., et al. (2019). Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun. 1:fcz022. doi: 10.1093/braincomms/fcz022
Santilli, F., Vazzana, N., Bucciarelli, L. G., and Davì, G. (2009). Soluble forms of RAGE in human diseases: clinical and therapeutical implications. Curr. Med. Chem. 16, 940–952. doi: 10.2174/092986709787581888
Sanz, J. M., Chiozzi, P., Ferrari, D., Colaianna, M., Idzko, M., Falzoni, S., et al. (2009). Activation of microglia by amyloid {beta} requires P2X7 receptor expression. J. Immunol. 182, 4378–4385. doi: 10.4049/jimmunol.0803612
Sardiello, M., Palmieri, M., di Ronza, A., Medina, D. L., Valenza, M., Gennarino, V. A., et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. doi: 10.1126/science.1174447
Sarlus, H., and Heneka, M. T. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249. doi: 10.1172/JCI90606
Saunders, A. M., Strittmatter, W. J., Schmechel, D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., et al. (1993). Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472. doi: 10.1212/wnl.43.8.1467
Saw, G., Krishna, K., Gupta, N., Soong, T. W., Mallilankaraman, K., Sajikumar, S., et al. (2020). Epigenetic regulation of microglial phosphatidylinositol 3-kinase pathway involved in long-term potentiation and synaptic plasticity in rats. Glia 68, 656–669. doi: 10.1002/glia.23748
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Scheiblich, H., Trombly, M., Ramirez, A., and Heneka, M. T. (2020). Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 41, 300–312. doi: 10.1016/j.it.2020.02.002
Scheuner, D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., et al. (1996). Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 2, 864–870. doi: 10.1038/nm0896-864
Schlepckow, K., Monroe, K. M., Kleinberger, G., Cantuti-Castelvetri, L., Parhizkar, S., Xia, D., et al. (2020). Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med. 12, e11227. doi: 10.15252/emmm.201911227
Schwartzentruber, J., Cooper, S., Liu, J. Z., Barrio-Hernandez, I., Bello, E., Kumasaka, N., et al. (2021). Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 53, 392–402. doi: 10.1038/s41588-020-00776-w
Sekino, S., Kashiwagi, Y., Kanazawa, H., Takada, K., Baba, T., Sato, S., et al. (2015). The NESH/Abi-3-based WAVE2 complex is functionally distinct from the Abi-1-based WAVE2 complex. Cell Commun. Signal. 13:41. doi: 10.1186/s12964-015-0119-5
Serrano-Pozo, A., Das, S., and Hyman, B. T. (2021). APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet. Neurol. 20, 68–80. doi: 10.1016/S1474-4422(20)30412-9
Sheedy, F. J., Grebe, A., Rayner, K. J., Kalantari, P., Ramkhelawon, B., Carpenter, S. B., et al. (2013). CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820. doi: 10.1038/ni.2639
Shen, Y., Lue, L., Yang, L., Roher, A., Kuo, Y., Strohmeyer, R., et al. (2001). Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 305, 165–168. doi: 10.1016/s0304-3940(01)01842-0
Shi, J.-Q., Zhang, C.-C., Sun, X.-L., Cheng, X.-X., Wang, J.-B., Zhang, Y.-D., et al. (2013). Antimalarial drug artemisinin extenuates amyloidogenesis and neuroinflammation in APPswe/PS1dE9 transgenic mice via inhibition of nuclear factor-κB and NLRP3 inflammasome activation. CNS Neurosci. Ther. 19, 262–268. doi: 10.1111/cns.12066
Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., et al. (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9:eaaf6295. doi: 10.1126/scitranslmed.aaf6295
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/JCI10498
Shibuya, Y., Niu, Z., Bryleva, E. Y., Harris, B. T., Murphy, S. R., Kheirollah, A., et al. (2015). Acyl-coenzyme A:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer’s disease mouse and reduces human P301L-tau content at the presymptomatic stage. Neurobiol. Aging 36, 2248–2259. doi: 10.1016/j.neurobiolaging.2015.04.002
Shieh, C.-H., Heinrich, A., Serchov, T., van Calker, D., and Biber, K. (2014). P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-α in cultured mouse microglia. Glia 62, 592–607. doi: 10.1002/glia.22628
Sica, A., and Mantovani, A. (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795. doi: 10.1172/JCI59643
Siegel, J. M., Schilly, K. M., Wijesinghe, M. B., Caruso, G., Fresta, C. G., and Lunte, S. M. (2019). Optimization of a microchip electrophoresis method with electrochemical detection for the determination of nitrite in macrophage cells as an indicator of nitric oxide production. Anal. Methods 11, 148–156. doi: 10.1039/C8AY02014K
Simard, A. R., Soulet, D., Gowing, G., Julien, J.-P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502. doi: 10.1016/j.neuron.2006.01.022
Sims, R., Hill, M., and Williams, J. (2020). The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 23, 311–322. doi: 10.1038/s41593-020-0599-5
Sims, R., van der Lee, S. J., Naj, A. C., Bellenguez, C., Badarinarayan, N., Jakobsdottir, J., et al. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384. doi: 10.1038/ng.3916
Slowik, A., Merres, J., Elfgen, A., Jansen, S., Mohr, F., Wruck, C. J., et al. (2012). Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE)–and amyloid beta 1-42-induced signal transduction in glial cells. Mol. Neurodegener. 7:55. doi: 10.1186/1750-1326-7-55
Solito, E., McArthur, S., Christian, H., Gavins, F., Buckingham, J. C., and Gillies, G. E. (2008). Annexin A1 in the brain–undiscovered roles? Trends Pharmacol. Sci. 29, 135–142. doi: 10.1016/j.tips.2007.12.003
Song, G. J., and Suk, K. (2017). Pharmacological modulation of functional phenotypes of microglia in neurodegenerative diseases. Front. Aging Neurosci. 9:139. doi: 10.3389/fnagi.2017.00139
Song, G. J., Nam, Y., Jo, M., Jung, M., Koo, J. Y., Cho, W., et al. (2016). A novel small-molecule agonist of PPAR-γ potentiates an anti-inflammatory M2 glial phenotype. Neuropharmacology 109, 159–169. doi: 10.1016/j.neuropharm.2016.06.009
Song, W., Hooli, B., Mullin, K., Jin, S. C., Cella, M., Ulland, T. K., et al. (2017). Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers. Dement. 13, 381–387. doi: 10.1016/j.jalz.2016.07.004
Sosna, J., Philipp, S., Albay, R. III, Reyes-Ruiz, J. M., Baglietto-Vargas, D., LaFerla, F. M., et al. (2018). Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 13:11. doi: 10.1186/s13024-018-0244-x
Spangenberg, E. E., Lee, R. J., Najafi, A. R., Rice, R. A., Elmore, M. R. P., Blurton-Jones, M., et al. (2016). Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 139, 1265–1281. doi: 10.1093/brain/aww016
Sperlágh, B., and Illes, P. (2014). P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol. Sci. 35, 537–547. doi: 10.1016/j.tips.2014.08.002
Srikanth, V., Maczurek, A., Phan, T., Steele, M., Westcott, B., Juskiw, D., et al. (2011). Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 32, 763–777. doi: 10.1016/j.neurobiolaging.2009.04.016
Stephan, A. H., Madison, D. V., Mateos, J. M., Fraser, D. A., Lovelett, E. A., Coutellier, L., et al. (2013). A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33, 13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013
Stewart, C. R., Stuart, L. M., Wilkinson, K., van Gils, J. M., Deng, J., Halle, A., et al. (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161. doi: 10.1038/ni.1836
Stoltzner, S. E., Grenfell, T. J., Mori, C., Wisniewski, K. E., Wisniewski, T. M., Selkoe, D. J., et al. (2000). Temporal accrual of complement proteins in amyloid plaques in Down’s syndrome with Alzheimer’s disease. Am. J. Pathol. 156, 489–499. doi: 10.1016/S0002-9440(10)64753-0
Su, F., Bai, F., Zhou, H., and Zhang, Z. (2016). Microglial toll-like receptors and Alzheimer’s disease. Brain. Behav. Immun. 52, 187–198. doi: 10.1016/j.bbi.2015.10.010
Suárez-Calvet, M., Araque Caballero, M. Á, Kleinberger, G., Bateman, R. J., Fagan, A. M., Morris, J. C., et al. (2016a). Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 8:369ra178. doi: 10.1126/scitranslmed.aag1767
Suárez-Calvet, M., Kleinberger, G., Araque Caballero, M. Á, Brendel, M., Rominger, A., Alcolea, D., et al. (2016b). sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med 8, 466–476. doi: 10.15252/emmm.201506123
Suárez-Calvet, M., Morenas-Rodríguez, E., Kleinberger, G., Schlepckow, K., Araque Caballero, M. Á, Franzmeier, N., et al. (2019). Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 14:1. doi: 10.1186/s13024-018-0301-5
Subhramanyam, C. S., Wang, C., Hu, Q., and Dheen, S. T. (2019). Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 94, 112–120. doi: 10.1016/j.semcdb.2019.05.004
Sun, D., Gao, G., Zhong, B., Zhang, H., Ding, S., Sun, Z., et al. (2021). NLRP1 inflammasome involves in learning and memory impairments and neuronal damages during aging process in mice. Behav. Brain Funct. 17:11. doi: 10.1186/s12993-021-00185-x
Sun, H., He, X., Tao, X., Hou, T., Chen, M., He, M., et al. (2020). The CD200/CD200R signaling pathway contributes to spontaneous functional recovery by enhancing synaptic plasticity after stroke. J. Neuroinflamm. 17:171. doi: 10.1186/s12974-020-01845-x
Takahashi, K., Rochford, C. D. P., and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657. doi: 10.1084/jem.20041611
Talebi, M., Delpak, A., Khalaj-Kondori, M., Sadigh-Eteghad, S., Talebi, M., Mehdizadeh, E., et al. (2020). ABCA7 and EphA1 genes polymorphisms in late-onset Alzheimer’s disease. J. Mol. Neurosci. 70, 167–173. doi: 10.1007/s12031-019-01420-x
Tang, Y., and Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Tao, Q.-Q., Chen, Y.-C., and Wu, Z.-Y. (2019). The role of CD2AP in the pathogenesis of Alzheimer’s Disease. Aging Dis. 10, 901–907. doi: 10.14336/AD.2018.1025
Tavana, J. P., Rosene, M., Jensen, N. O., Ridge, P. G., Kauwe, J. S., and Karch, C. M. (2019). RAB10: an Alzheimer’s disease resilience locus and potential drug target. Clin. Interv. Aging 14, 73–79. doi: 10.2147/CIA.S159148
Tenner, A. J. (2001). Complement in Alzheimer’s disease: opportunities for modulating protective and pathogenic events. Neurobiol. Aging 22, 849–861. doi: 10.1016/s0197-4580(01)00301-3
Territo, P. R., Meyer, J. A., Peters, J. S., Riley, A. A., McCarthy, B. P., Gao, M., et al. (2017). Characterization of (11)C-GSK1482160 for targeting the P2X7 receptor as a biomarker for neuroinflammation. J. Nucl. Med. 58, 458–465. doi: 10.2967/jnumed.116.181354
Thawkar, B. S., and Kaur, G. (2019). Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 326, 62–74. doi: 10.1016/j.jneuroim.2018.11.010
Therriault, J., Benedet, A. L., Pascoal, T. A., Mathotaarachchi, S., Chamoun, M., Savard, M., et al. (2020). Association of apolipoprotein E ε4 with medial temporal tau independent of Amyloid-β. JAMA Neurol. 77, 470–479. doi: 10.1001/jamaneurol.2019.4421
Tortora, F., Rendina, A., Angiolillo, A., Di Costanzo, A., Aniello, F., Donizetti, A., et al. (2022). CD33 rs2455069 SNP: correlation with Alzheimer’s Disease and Hypothesis of Functional Role. Int. J. Mol. Sci. 23:3629. doi: 10.3390/ijms23073629
Tóth, B., Garabuczi, E., Sarang, Z., Vereb, G., Vámosi, G., Aeschlimann, D., et al. (2009). Transglutaminase 2 is needed for the formation of an efficient phagocyte portal in macrophages engulfing apoptotic cells. J. Immunol. 182, 2084–2092. doi: 10.4049/jimmunol.0803444
Tremblay, M. -È, Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Tsai, A. P., Dong, C., Lin, P. B.-C., Messenger, E. J., Casali, B. T., Moutinho, M., et al. (2022). PLCG2 is associated with the inflammatory response and is induced by amyloid plaques in Alzheimer’s disease. Genome Med. 14:17. doi: 10.1186/s13073-022-01022-0
Tsai, A. P., Lin, P. B.-C., Dong, C., Moutinho, M., Casali, B. T., Liu, Y., et al. (2021). INPP5D expression is associated with risk for Alzheimer’s disease and induced by plaque-associated microglia. Neurobiol. Dis. 153:105303. doi: 10.1016/j.nbd.2021.105303
Ullman, J. C., Arguello, A., Getz, J. A., Bhalla, A., Mahon, C. S., Wang, J., et al. (2020). Brain delivery and activity of a lysosomal enzyme using a blood-brain barrier transport vehicle in mice. Sci. Transl. Med. 12:eaay1163. doi: 10.1126/scitranslmed.aay1163
Ulrich, J. D., Finn, M. B., Wang, Y., Shen, A., Mahan, T. E., Jiang, H., et al. (2014). Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 9:20. doi: 10.1186/1750-1326-9-20
Ulrich, J. D., Ulland, T. K., Mahan, T. E., Nyström, S., Nilsson, K. P., Song, W. M., et al. (2018). ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 215, 1047–1058. doi: 10.1084/jem.20171265
Vainchtein, I. D., Chin, G., Cho, F. S., Kelley, K. W., Miller, J. G., Chien, E. C., et al. (2018). Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273. doi: 10.1126/science.aal3589
Van Zeller, M., Sebastião, A. M., and Valente, C. A. (2022). Microglia depletion from primary glial cultures enables to accurately address the immune response of astrocytes. Biomolecules 12:666. doi: 10.3390/biom12050666
Vardarajan, B. N., Barral, S., Jaworski, J., Beecham, G. W., Blue, E., Tosto, G., et al. (2018). Whole genome sequencing of caribbean hispanic families with late-onset Alzheimer’s disease. Ann. Clin. Transl. Neurol. 5, 406–417. doi: 10.1002/acn3.537
Végh, M. J., Heldring, C. M., Kamphuis, W., Hijazi, S., Timmerman, A. J., Li, K. W., et al. (2014a). Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2:76. doi: 10.1186/s40478-014-0076-z
Végh, M. J., Rausell, A., Loos, M., Heldring, C. M., Jurkowski, W., van Nierop, P., et al. (2014b). Hippocampal extracellular matrix levels and stochasticity in synaptic protein expression increase with age and are associated with age-dependent cognitive decline. Mol. Cell. Proteomics 13, 2975–2985. doi: 10.1074/mcp.M113.032086
Verghese, P. B., Castellano, J. M., Garai, K., Wang, Y., Jiang, H., Shah, A., et al. (2013). ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U S A. 110, E1807–E1816. doi: 10.1073/pnas.1220484110
von Bernhardi, R., Eugenín-von Bernhardi, L., and Eugenín, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
Wahrle, S. E., Jiang, H., Parsadanian, M., Hartman, R. E., Bales, K. R., Paul, S. M., et al. (2005). Deletion of abca1 increases abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242. doi: 10.1074/jbc.M508780200
Wang, C., Najm, R., Xu, Q., Jeong, D.-E., Walker, D., Balestra, M. E., et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. doi: 10.1038/s41591-018-0004-z
Wang, C., Xiong, M., Gratuze, M., Bao, X., Shi, Y., Andhey, P. S., et al. (2021). Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109, 1657–1674.e7. doi: 10.1016/j.neuron.2021.03.024.
Wang, C., Yue, H., Hu, Z., Shen, Y., Ma, J., Li, J., et al. (2020). Microglia mediate forgetting via complement-dependent synaptic elimination. Science 367, 688–694. doi: 10.1126/science.aaz2288
Wang, J., Xing, H., Wan, L., Jiang, X., Wang, C., and Wu, Y. (2018). Treatment targets for M2 microglia polarization in ischemic stroke. Biomed. Pharmacother. 105, 518–525. doi: 10.1016/j.biopha.2018.05.143
Wang, S., Mustafa, M., Yuede, C. M., Salazar, S. V., Kong, P., Long, H., et al. (2020). Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 217:e20200785. doi: 10.1084/jem.20200785
Wang, X.-H., Xie, X., Luo, X.-G., Shang, H., and He, Z.-Y. (2017). Inhibiting purinergic P2X7 receptors with the antagonist brilliant blue G is neuroprotective in an intranigral lipopolysaccharide animal model of Parkinson’s disease. Mol. Med. Rep. 15, 768–776. doi: 10.3892/mmr.2016.6070
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675. doi: 10.1084/jem.20151948
Wang, Y.-J., Monteagudo, A., Downey, M. A., Ashton-Rickardt, P. G., and Elmaleh, D. R. (2021). Cromolyn inhibits the secretion of inflammatory cytokines by human microglia (HMC3). Sci. Rep. 11:8054. doi: 10.1038/s41598-021-85702-8
Wang, Y.-T. T., Pascoal, T. A., Therriault, J., Kang, M. S., Benedet, A. L., Savard, M., et al. (2021). Interactive rather than independent effect of APOE and sex potentiates tau deposition in women. Brain Commun. 3:fcab126. doi: 10.1093/braincomms/fcab126
Webster, S. D., Park, M., Fonseca, M. I., and Tenner, A. J. (2000). Structural and functional evidence for microglial expression of C1qR(P), the C1q receptor that enhances phagocytosis. J. Leukoc. Biol. 67, 109–116. doi: 10.1002/jlb.67.1.109
Weigand, A. J., Thomas, K. R., Bangen, K. J., Eglit, G. M. L., Delano-Wood, L., Gilbert, P. E., et al. (2021). APOE interacts with tau PET to influence memory independently of amyloid PET in older adults without dementia. Alzheimers. Dement. 17, 61–69. doi: 10.1002/alz.12173
Weinhard, L., di Bartolomei, G., Bolasco, G., Machado, P., Schieber, N. L., Neniskyte, U., et al. (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 9:1228. doi: 10.1038/s41467-018-03566-5
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266. doi: 10.1111/j.1750-3639.2008.00133.x
Wilkinson, K., and El Khoury, J. (2012). Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer’s disease. Int. J. Alzheimers. Dis. 2012:489456. doi: 10.1155/2012/489456
Wilkinson, K., Boyd, J. D., Glicksman, M., Moore, K. J., and El Khoury, J. (2011). A high content drug screen identifies ursolic acid as an inhibitor of amyloid beta protein interactions with its receptor CD36. J. Biol. Chem. 286, 34914–34922. doi: 10.1074/jbc.M111.232116
Williams, T., Borchelt, D. R., and Chakrabarty, P. (2020). Therapeutic approaches targeting apolipoprotein E function in Alzheimer’s disease. Mol. Neurodegener. 15:8. doi: 10.1186/s13024-020-0358-9
Wolfe, M. S. (2008). Gamma-secretase: structure, function, and modulation for Alzheimer’s disease. Curr. Top. Med. Chem. 8, 2–8. doi: 10.2174/156802608783334024
Woods, L. T., Ajit, D., Camden, J. M., Erb, L., and Weisman, G. A. (2016). Purinergic receptors as potential therapeutic targets in Alzheimer’s disease. Neuropharmacology 104, 169–179. doi: 10.1016/j.neuropharm.2015.10.031
Worthen, R. J., Garzon Zighelboim, S. S., Torres Jaramillo, C. S., and Beurel, E. (2020). Anti-inflammatory IL-10 administration rescues depression-associated learning and memory deficits in mice. J. Neuroinflamm. 17:246. doi: 10.1186/s12974-020-01922-1
Wu, F., Luo, T., Mei, Y., Liu, H., Dong, J., Fang, Y., et al. (2018). Simvastatin alters M1/M2 polarization of murine BV2 microglia via Notch signaling. J. Neuroimmunol. 316, 56–64. doi: 10.1016/j.jneuroim.2017.12.010
Wu, K., Byers, D. E., Jin, X., Agapov, E., Alexander-Brett, J., Patel, A. C., et al. (2015). TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med. 212, 681–697. doi: 10.1084/jem.20141732
Wunderlich, P., Glebov, K., Kemmerling, N., Tien, N. T., Neumann, H., and Walter, J. (2013). Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33027–33036. doi: 10.1074/jbc.M113.517540
Wyss-Coray, T., and Rogers, J. (2012). Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2:a006346. doi: 10.1101/cshperspect.a006346
Xin, P., Xu, X., Deng, C., Liu, S., Wang, Y., Zhou, X., et al. (2020). The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 80:106210. doi: 10.1016/j.intimp.2020.106210
Xu, Y., Xu, Y., Wang, Y., Wang, Y., He, L., Jiang, Z., et al. (2015). Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKβ-dependent AMPK activation. Brain. Behav. Immun. 50, 298–313. doi: 10.1016/j.bbi.2015.07.015
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C.-C., and Bu, G. (2019). Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518. doi: 10.1038/s41582-019-0228-7
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., et al. (1996). RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691. doi: 10.1038/382685a0
Yan, S. F., Ramasamy, R., and Schmidt, A. M. (2009). Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J. Mol. Med. (Berl). 87, 235–247. doi: 10.1007/s00109-009-0439-2
Yancey, P. G., Blakemore, J., Ding, L., Fan, D., Overton, C. D., Zhang, Y., et al. (2010). Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and Akt activation. Arterioscler. Thromb. Vasc. Biol. 30, 787–795. doi: 10.1161/ATVBAHA.109.202051
Yang, C.-N., Shiao, Y.-J., Shie, F.-S., Guo, B.-S., Chen, P.-H., Cho, C.-Y., et al. (2011). Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiol. Dis. 42, 221–230. doi: 10.1016/j.nbd.2011.01.005
Yang, Y., Wang, H., Kouadir, M., Song, H., and Shi, F. (2019). Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 10:128. doi: 10.1038/s41419-019-1413-8
Yang, Z., Liu, B., Yang, L.-E., and Zhang, C. (2019). Platycodigenin as potential drug candidate for Alzheimer’s disease via modulating microglial polarization and neurite regeneration. Molecules 24:3207. doi: 10.3390/molecules24183207
Yin, J., Zhao, F., Chojnacki, J. E., Fulp, J., Klein, W. L., Zhang, S., et al. (2018). NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 55, 1977–1987. doi: 10.1007/s12035-017-0467-9
Yona, S., Buckingham, J. C., Perretti, M., and Flower, R. J. (2004). Stimulus-specific defect in the phagocytic pathways of annexin 1 null macrophages. Br. J. Pharmacol. 142, 890–898. doi: 10.1038/sj.bjp.0705858
Young, K. A., Hirst, W. D., Solito, E., and Wilkin, G. P. (1999). De novo expression of lipocortin-1 in reactive microglia and astrocytes in kainic acid lesioned rat cerebellum. Glia 26, 333–343. doi: 10.1002/(sici)1098-1136(199906)26:4<333::aid-glia7>3.0.co;2-s
Yu, M., Qi, B., Xiaoxiang, W., Xu, J., and Liu, X. (2017). Baicalein increases cisplatin sensitivity of A549 lung adenocarcinoma cells via PI3K/Akt/NF-κB pathway. Biomed. Pharmacother. 90, 677–685. doi: 10.1016/j.biopha.2017.04.001
Yu, Y., and Ye, R. D. (2015). Microglial Aβ receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 35, 71–83. doi: 10.1007/s10571-014-0101-6
Yuan, J., Amin, P., and Ofengeim, D. (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33. doi: 10.1038/s41583-018-0093-1
Zhang, D.-W., Shao, J., Lin, J., Zhang, N., Lu, B.-J., Lin, S.-C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. doi: 10.1126/science.1172308
Zhang, H., Li, Y., Yu, J., Guo, M., Meng, J., Liu, C., et al. (2013). Rho kinase inhibitor fasudil regulates microglia polarization and function. Neuroimmunomodulation 20, 313–322. doi: 10.1159/000351221
Zhang, Y., Shen, L., Zhu, H., Dreissigacker, K., Distler, D., Zhou, X., et al. (2020). PGC-1α regulates autophagy to promote fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 79, 1227–1233. doi: 10.1136/annrheumdis-2020-216963
Zhao, J., Fu, Y., Yamazaki, Y., Ren, Y., Davis, M. D., Liu, C.-C., et al. (2020a). APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 11:5540. doi: 10.1038/s41467-020-19264-0
Zhao, N., Li, C.-C., Di, B., and Xu, L.-L. (2020b). Recent advances in the NEK7-licensed NLRP3 inflammasome activation: mechanisms, role in diseases and related inhibitors. J. Autoimmun. 113:102515. doi: 10.1016/j.jaut.2020.102515
Zhao, N., Ren, Y., Yamazaki, Y., Qiao, W., Li, F., Felton, L. M., et al. (2020c). Alzheimer’s risk factors age, APOE genotype, and sex drive distinct molecular pathways. Neuron 106, 727–742.e6. doi: 10.1016/j.neuron.2020.02.034.
Zhao, Q.-F., Yu, J.-T., Tan, M.-S., and Tan, L. (2015). ABCA7 in Alzheimer’s disease. Mol. Neurobiol. 51, 1008–1016. doi: 10.1007/s12035-014-8759-9
Zhao, W., Zhang, J., Davis, E. G., and Rebeck, G. W. (2014). Aging reduces glial uptake and promotes extracellular accumulation of Aβ from a lentiviral vector. Front. Aging Neurosci. 6:210. doi: 10.3389/fnagi.2014.00210
Zhao, Y., Cong, L., Jaber, V., and Lukiw, W. J. (2017a). Microbiome-Derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front. Immunol. 8:1064. doi: 10.3389/fimmu.2017.01064
Zhao, Y., Jaber, V., and Lukiw, W. J. (2017b). Secretory products of the human gi tract microbiome and their potential impact on Alzheimer’s Disease (AD): detection of lipopolysaccharide (LPS) in AD hippocampus. Front. Cell. Infect. Microbiol. 7:318. doi: 10.3389/fcimb.2017.00318
Zhao, Y., Wu, X., Li, X., Jiang, L.-L., Gui, X., Liu, Y., et al. (2018). TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023–1031.e7. doi: 10.1016/j.neuron.2018.01.031
Zhong, L., Chen, X.-F., Wang, T., Wang, Z., Liao, C., Wang, Z., et al. (2017). Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 214, 597–607. doi: 10.1084/jem.20160844
Zhong, L., Xu, Y., Zhuo, R., Wang, T., Wang, K., Huang, R., et al. (2019). Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 10:1365. doi: 10.1038/s41467-019-09118-9
Zhu, H., Wang, Z., Yu, J., Yang, X., He, F., Liu, Z., et al. (2019). Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog. Neurobiol. 178:101610. doi: 10.1016/j.pneurobio.2019.03.003
Zhu, X.-C., Yu, J.-T., Jiang, T., Wang, P., Cao, L., and Tan, L. (2015). CR1 in Alzheimer’s disease. Mol. Neurobiol. 51, 753–765. doi: 10.1007/s12035-014-8723-8
Zirngibl, M., Assinck, P., Sizov, A., Caprariello, A. V., and Plemel, J. R. (2022). Oligodendrocyte death and myelin loss in the cuprizone model: an updated overview of the intrinsic and extrinsic causes of cuprizone demyelination. Mol. Neurodegener. 17:34. doi: 10.1186/s13024-022-00538-8
Zorzetto, M., Datturi, F., Divizia, L., Pistono, C., Campo, I., De Silvestri, A., et al. (2017). Complement C4A and C4B gene copy number study in Alzheimer’s disease patients. Curr. Alzheimer Res. 14, 303–308. doi: 10.2174/1567205013666161013091934
Zotova, E., Bharambe, V., Cheaveau, M., Morgan, W., Holmes, C., Harris, S., et al. (2013). Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain 136, 2677–2696. doi: 10.1093/brain/awt210
Glossary
| Abbreviation | Full name |
| ABCA7 | ATP-binding cassette sub-family A member 7 |
| ACT | Antichymotrypsin |
| AD | Alzheimer’s disease |
| ADAM | A disintegrin and metalloprotease |
| ADO | Adenosine |
| ADSP | AD Sequencing Project |
| AKAP9 | A-kinase anchor protein 9 |
| ALS | Amyotrophic lateral sclerosis |
| ANXA1 | Annexin A1 |
| AP-1 | Activator protein 1 |
| APP | Amyloid precursor protein |
| Arg-1 | Arginase 1 |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid beta |
| BAI1 | Brain-specific angiogenesis inhibitor 1 |
| BBB | Blood-brain barrier |
| BBG | Brilliant blue G |
| BDNF | Brain-derived neurotrophic factor |
| BIN1 | Bridging integrator 1 |
| BITC | Benzyl isothiocyanate |
| C2CD3 | C2 Domain Containing 3 Centriole Elongation Regulator |
| CaMKKβ | Calcium/calmodulin-dependent protein kinase kinase-β |
| CAR-Ps | Chimeric antigen receptor-phagocytic receptors |
| CD33 | Siglec-3 |
| CLEAR | Coordinated lysosomal expression and regulation |
| CLSTN2 | Calsyntenin 2 |
| CNS | Central Nervous System |
| CR1 | Complement receptor 1 |
| CTNNA2 | N-catenin and Catenin alpha-2 |
| CX3CR1 | CX3C chemokine receptor 1 |
| DAM | Disease-associated microglia |
| DAMPs | Danger-associated molecular patterns |
| EAE | Experimental autoimmune encephalomyelitis |
| ECM | Extracellular matrix |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| EXOC5 | Exocyst complex component 5 |
| FERMT2 | Fermitin family homolog 2 |
| FNBP1L | Formin-binding protein 1-like |
| GAS2L2 | GAS2-like protein 2 |
| Gas6 | Growth arrest-specific protein 6 |
| GPCRs | G-protein-coupled receptors |
| GULP1 | PTB domain-containing engulfment adapter protein 1 |
| GWAS | Genome-wide association studies |
| HO-1 | Heme oxygenase-1 |
| IκB | Inhibitors of NF-κB |
| IKK | IκB kinase |
| IL-4 | Interleukin 4 |
| IRAK | Interleukin-1 receptor-associated kinase |
| JAK/STAT | Janus kinase/signal transducer and activator of transcription |
| KIF2A | Kinesin heavy chain member 2A |
| LAP | LC3-associated phagocytosis |
| LHX9 | LIM Homeobox 9 |
| LKB1 | Liver kinase B1 |
| LOAD | Late-onset Alzheimer’s Disease |
| LPC | Lipocortin |
| LPS | Lipopolysaccharide |
| LRP1 | LDL-receptor-associated protein 1 |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MAPK | Mitogen-activated protein kinases |
| MFG-E8 | Milk fat globule-EGF factor 8 protein |
| MS | Multiple sclerosis |
| MyD88 | Myeloid differentiation factor 88 |
| NALCN | Sodium Leak Channel, Non-Selective |
| NDRG2 | NDRG Family Member 2 |
| NF-κB | Nuclear factor-κB |
| NFTs | Neurofibrillary tangles |
| NICD | Notch intracellular domain |
| NLRs | Nucleotide-binding oligomerization domain-like receptors |
| NO | Nitric oxide |
| NOX | Nicotinamide adenine dinucleotide phosphate oxidase |
| NSF | Vesicle-fusing ATPase |
| OECs | Olfactory ensheathing cells |
| P2 × 7R | P2 × 7 receptor |
| PAMP | Pathogen-associated molecular patterns |
| PILRA | Paired immunoglobin-like type 2 receptor alpha |
| PI3K | Phosphoinositide 3-kinase |
| PGC-1α | Peroxisome proliferator-activated receptor γ coactivator 1α |
| PKB | Protein kinase B |
| PLC | Phospholipase C |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| PRKCH | Protein kinase C |
| PRRs | Pattern recognition receptors |
| PSD-95 | Postsynaptic density protein-95 |
| PSEN1 | Presenilin 1 |
| PtdSer | Phosphatidylserine |
| RAGE | Receptor for advanced glycation end products |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RIPK1 | Receptor-interacting serine/threonine kinase 1 |
| ROS | Reactive oxygen species |
| RXRs | Retinoid X receptors |
| S1P | Sphingosine-1-phosphate |
| SAP | Serum amyloid protein |
| SAβ | Soluble amyloid beta |
| SEL1L | Suppressor of lin-12-like protein 1 |
| sFKN | Soluble fractalkine |
| SIGLECs | Sialic acid-binding immunoglobulin-like lectins |
| SIRPα | Signal-regulatory protein alpha |
| Sirt1 | Sirtuin 1 |
| SLC24A4 | Solute carrier family 24 member 4 |
| SNP | Single nucleotide polymorphism |
| SOCS | Suppressor of cytokine signaling |
| SORL1 | Sortilin related receptor 1 |
| SRA | Scavenger receptor A |
| SR-A1 | Class A1 scavenger receptors |
| SR-MARCO | Scavenger receptor and macrophage receptor with collagenous structure |
| STAT | Signal Transducer and Activator of Transcription |
| STC | Synaptic tagging and capture |
| SYTL3 | Synaptotagmin-like 3 |
| TAM | Tyrosine kinases |
| TAK1 | Transforming growth factor-β-activated kinase-1 |
| TDP-43 | 43 kDa TARDNA binding protein |
| TFEB | Transcription factor EB |
| TFEC | Transcription factor EC |
| MITF | Microphthalmia-transcription-factor |
| TG2 | Transglutaminase 2 |
| TIAF1 | TGFB1-induced antiapoptotic factor 1 |
| TLR s | Toll-like receptors |
| TMEM132A | Transmembrane Protein 132A |
| TNF | Tumor necrosis factor |
| TRAF6 | Tumor necrosis factor receptor associated factor-6 |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| VR | Vitronectin receptor |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| WNT7A | Wnt Family Member 7A |
| ZNF655 | Zinc Finger Protein 655 |
Keywords: Alzheimer’s disease, microglia, amyloid-beta, phagocytosis, neuroinflammation
Citation: Miao J, Ma H, Yang Y, Liao Y, Lin C, Zheng J, Yu M and Lan J (2023) Microglia in Alzheimer’s disease: pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 15:1201982. doi: 10.3389/fnagi.2023.1201982
Received: 07 April 2023; Accepted: 30 May 2023;
Published: 15 June 2023.
Edited by:
Xuemin Xu, University of Texas of the Permian Basin, United StatesReviewed by:
Ke Zhang, China Medical University, ChinaBen J. Gu, University of Melbourne, Australia
Copyright © 2023 Miao, Ma, Yang, Liao, Lin, Zheng, Yu and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiao Lan, 505970191@qq.com